FiO2 0.5 via a non re-breathe reservoir bag/mask
Arterial blood gas (ABG) analysis:
pH 7.3
PaO2 11 kPa
PaCO2 6.9 kPa
Base Excess −4
Heart rate 69 beats per minute, sinus rhythm
Capillary refill < 2 seconds
Pupils are unequal – left = 5 mm, right = 3 m. The right pupil reacts during testing of direct and consensual light reflexes; the left pupil remains fixed.
Blood glucose 4.9 mmol/L
Large scalp laceration over the left-temporal region and bruising over the left side of his face
No other apparent injuries, although a cervical collar is in situ as a precaution for spinal injury.
Reader activities
Having read the scenario, consider the following:
- What is the cause of Paul’s rapid neurological deterioration?
- What are the physiological mechanisms that maintain normal cerebral blood flow (CBF) and ICP?
- What are the implications for Paul of a raised ICP?
- What physiological changes would be evident with a rising ICP?
Pathophysiology related to increased ICP
The skull is a rigid box structure with incompressible contents of brain tissue, cerebral spinal fluid (CSF) and blood, which exist in a state of volume equilibrium. An increase in the volume of one constituent must be compensated by a decrease in the volume of another – the ‘Monro-Kellie doctrine’ (Lindsay et al. 2004; Lettieri 2006). Compromises in intracranial compliance or an expanding mass within the skull will consequently increase ICP (Lettieri 2006). Usually increases in ICP are controlled by autoregulatory mechanisms such as the displacement of blood via venous drainage or CSF into the spinal compartment/lumbar theca. These compensatory mechanisms will maintain ICP within physiological parameters (0–15 mmHg) up to a physiological limit (Lettieri 2006).
Maintenance of CBF is dependent on cerebral vascular resistance and cerebral perfusion pressure (CPP). CPP is calculated by subtracting mean ICP from mean arterial blood pressure (MABP) (Arbour 2004). An adequate CPP is vital in the maintenance of global cerebral perfusion and prevention of cerebral ischaemia. A CPP of above 70 mmHg is generally viewed as desirable to maintain adequate perfusion, but in TBI levels of between 60 and 70 mmHg may be acceptable (Arbour 2004). CBF is also coupled to the metabolic and energy requirements of brain tissue (Lindsay et al. 2004); essentially the rate is approximately 50 mL/100 mg/min (Ridley et al. 2003).
Following severe traumatic brain injury (TBI) autoregulation of CBF is impaired (Lindsay et al. 2004). Initially, cerebral blood volume (CBV) increases due to cerebral vasodilatation caused by raised carbon dioxide levels (PaCO2), which decreases extracellular pH provoking a rise in ICP. Sustained raised ICP reduces CPP compromising CBF, and decreasing CBV. Reduced cerebral perfusion of oxygen sensitive cells in the cortex causes anaerobic cell metabolism leading to ischaemia and a build-up of metabolic by-products that lower extracellular pH (Layton et al. 2004). When CBF is < 25 mL/100 mg/min electrical activity is severely compromised and if the rate decreases below 10 mL/100 mg/min cell death occurs (Ridley et al. 2003). Hypoxia, ischaemia, low extracellular pH and blood hypo-osmolality lead to cytotoxic oedema – intracellular fluid accumulation. This compounds vasogenic oedema, which is believed to occur soon after injury and results from disruption of the blood–brain barrier (BBB), causing increased permeability of capillary walls and leakage of protein- rich fluid into the extracellular space (Ridley et al. 2003). Development of oedema increases ICP, and eventually compensatory mechanisms are exhausted. In a congested brain, CBF becomes compromised and increased pressure on the vascular bed leads to distortion and increased vascular resistance, which restricts cerebral perfusion perpetuating a vicious cycle of oedema formation and raised ICP (Lindsay et al. 2004). Raised ICP can also be caused by an expanding haematoma, restricted venous outflow and obstruction to CSF flow and /or absorption (Singh and Stock 2006). Any expanding mass causes brain tissue to shift risking herniation across fixed dural structures within the brain, leading to irreversible and potentially fatal damage (Arbour 2004; Shepard 2004).
Paul’s increasing ICP and initial hypoxia would cause loss of consciousness, which compromises the airway (Lettieri 2006). Raised ICP causes pressure on the respiratory centres of the medulla oblongata in the brainstem altering breathing rate and pattern – often hypoventilation or an irregular breathing with apnoeic episodes (Mayer and Chong 2002). Inadequate ventilation may lead to a raised PaCO2, which causes cerebral vasodilatation, further increasing the ICP (Mayer and Chong 2002).
Paul was diagnosed with a sub-dural haematoma. An expanding lesion on one side of the brain may cause haemiparesis/haemiplegia due to direct injury or pressure on the motor cortex (Shepard 2004). Nerve impulses for voluntary movement originate in the motor cortex of the frontal lobe (Tortora and Grabowski 2000). Upper motor neurones travel via pyramidal pathways, most ‘cross over’ or decussate at the level of the medulla oblongata in the brainstem, before descending the spinal cord to synapse with lower motor neurones that innervate skeletal muscle of the trunk and limbs (Tortora and Grabowski 2000). Hence, Paul’s injury to the left motor cortex will cause a contralateral (right-sided) haemiparesis/haemiplegia (Lindsay et al. 2004).
As Paul’s ICP increases, assessment of his pupils reveals altered size and reaction to light, which is due to medial temporal lobe pressure on the oculomotor nerve (cranial nerve III) (Lindsay et al. 2004). Initially oculomotor nerve damage is ipsilateral but may become bilateral with increasing ICP. On fundoscopy, papilloedema may be evident if increased CSF pressure in the optic nerve (cranial nerve II) sheath has impeded venous drainage, leading to retinal and optic disc haemorrhages (Lindsay et al. 2004). Generally, raised ICP causes a persistent headache due to stretching of the arteries in the brainstem and displacement of CSF. Vomiting is a frequent side effect due to pressure on the ‘vomit’ centre of the brainstem (Singh and Stock. 2006).
Factors that instigate changes in ICP cannot be considered separately because they each contribute to complex inter-relationships or feedback pathways that can actually exacerbate further brain injury (Lindsay et al. 2004).
In TBI, intracranial bleeding can be extradural, subdural or intracerebral; sometimes sub-arachnoid bleeding may also occur (Lettieri 2006). Secondary brain injury such as haematomas, ischaemia, infection, brain swelling and shift occur after the initial primary injury (Shepard 2004). Restricting the impact of secondary causes on ICP is the focus for management of patients with TBI.
Developing scenario
Paul arrives at the Neurosurgical Centre and immediately undergoes surgery to remove the subdural haematoma. He is transferred to the intensive care unit post-surgery.
Reader activities
Consider the following:
- What would be the main aims of treatment to limit the impact of Paul’s raised ICP? (Consider the relationship between CBF, CPP and ICP. Review other monitoring modalities and their value in managing TBI).
- Paul is sedated and ventilated: What would be the optimal parameters for arterial blood gases for a patient with a raised ICP?
- How might you assess Paul’s neurological function whilst he is sedated?
- What other nursing considerations will there be during the first 48 hours post-surgery?
Assessment tools
The injured brain is exceptionally susceptible to secondary ischaemic insults such as hypoxia and hypotension, which are associated with poor neurological outcome after severe TBI (Hlatky et al. 2003). Management of patients with TBI is therefore frequently dominated by techniques based on maintaining cerebral perfusion and adequacy of CBF with concomitant limitation of increases in ICP (Hlatky et al. 2003; Young et al. 2003).
Maintenance of Paul’s CPP is important because of the significant physiological role it represents in a patient with TBI. CPP indicates the pressure gradient acting across the cerebrovascular bed, and therefore it is an important factor in the regulation of CBF (Hlatky et al. 2003). CPP contributes to the hydrostatic pressure within intracerebral vessels, so it has the potential to affect oedema formation in the injured brain (Hlatky et al. 2003).
Monitoring of Paul’s ICP to determine his CPP warrants insertion of both an arterial line and ICP monitoring device. Monitoring of ICP has been shown to be useful in predicting outcome from TBI and in guiding therapy for raised ICP, which may lead to improved outcome for those patients who respond to ICP lowering interventions (Brain Trauma Foundation (BTF) 2007). A number of devices are currently available for the monitoring of ICP, including insertion of an intraventricular catheter, subarachnoid bolt or screw, or a subdural or epidural catheter and intraparenchymal insertion of a fiber-optic transducer tipped catheter (see Figure 12.1). A good overview of the range of ICP assessment methods and their advantages and disadvantages can be found in Arbour (2004).
Figure 12.1 Major types of intracranial pressure monitoring. Coronal section of the brain shows potential sites for placement of the monitoring devices. (Adapted from Kerr and Crago, with permission from Elsevier.)
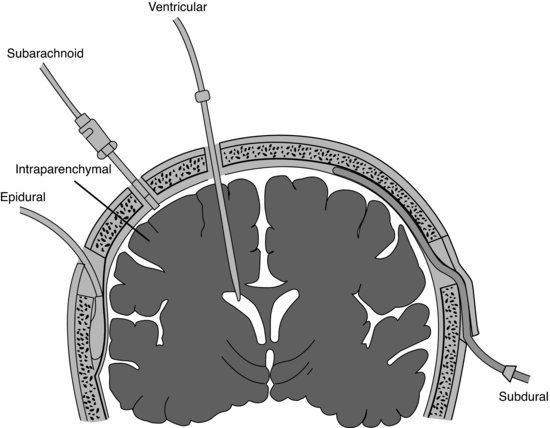
Intraventricular devices are currently viewed as the gold standard for accurate ICP monitoring (Arbour 2004; BTF 2007). There is a therapeutic advantage to inserting an intraventricular catheter because it allows for highly accurate measurement of ICP and also allows for the drainage of CSF if necessary (Lettieri 2006; BTF 2007). Intraventricular devices are, however, associated with a higher incidence of infection compared to an intraparenchymal microtransducer device (e.g. Codman® ICP probes) (Lane et al. 2000). Less invasive devices provide less accurate measurements and may require frequent recalibrations (Arbour 2004).
In patients with TBI, the CPP should be maintained above 60 mmHg (Juul et al. 2000; Arbour 2004). A CPP of 55 mmHg is considered a probable critical threshold for poor neurological outcome in adults and a CPP of < 50 mmHg should be avoided (BTF 2007); however, it is important to remember that CPP may be non-uniform with variable CBF to areas of injury (Lettieri 2006). Mindful of this and allowing for variations in MABP, most clinicians prefer to maintain CPP within a target range of 50–70 mmHg (BTF 2007). Aggressive attempts to maintain CPP above 70 mmHg with fluids and vasoactive drugs should be avoided because of the risk of adult respiratory distress syndrome (ARDS) (BTF 2007).
Measurements of cerebral oxygenation are also recommended in the patient with TBI (BTF 2007), and evidence supports the use of either jugular venous saturations (SjvO2) or brain tissue oxygen levels (PbtO2) Measurements indicate the adequacy of CBF relative to metabolic requirements; for example, with rising ICP, resultant hypoperfusion will increase oxygen extraction and SJvO2 will be decreased <50% (normal SJvO2 55 – 75%) (Hlatky et al. 2003). The major limitation of using SJvO2 as a monitor of CBF adequacy is that regional ischaemia will not be identified (Hlatky et al. 2003; Layton et al. 2004). In TBI, where regional differences in CBF may occur, monitoring of brain tissue PbtO2 may be beneficial. Normal values are approximately 20–40 mmHg with a range of 8–10 mmHg representing a probable critical threshold (Hoffman et al. 1996; Kiening et al. 1996; Valadka et al. 1998, 2000; BTF 2007).
Managing the patient with raised ICP
Nursing considerations for critically ill brain injured patients aim to optimise systemic and cerebral oxygenation and CBF and minimise factors that may aggravate neuronal injury or contribute to intracranial hypertension (Mayer and Chong 2002).
Airway/breathing
If the GCS is ≤8 intubation and ventilation are required to protect the airway, allow for deep sedation, and to facilitate optimal gas exchange in the lungs. In patients with TBI, optimal oxygenation and prevention of hypoxaemia are paramount. Ventilation manipulation should aim to maintain arterial oxygen (PaO2) levels above 12 kPa, with strenuous avoidance of PaO2 <8.0 mmHg or O2 saturation <90% (BTF 2007). Arterial CO2 (PaCO2) levels should be maintained at between 4.5 and 5.0 kPa. Hypercarbia (PaCO2 >6 kPa) is avoided because it can cause cerebral vasodilatation and hyperaemia, which increase ICP (Lettieri 2006). Therapeutic hyperventilation, however, produces hypocarbia, which causes cerebral vasoconstriction, decreasing CBV and hence ICP (Yanko and Mitcho 2001). Even so, CBF generally decreases during the initial 24 hours following severe TBI and vasoresponsivity to carbon dioxide may therefore be reduced negating the positive effects of vasoconstriction; coupled with a loss of autoregulation, ICP may not actually decrease (Lettieri 2006). Increasing the rate of CO2 removal from the blood will decrease hydrogen ion concentration in CSF, which precipitates chemical and ionic imbalances, such as alkalosis and hypokaleamia (Yanko and Mitcho 2001). Mayer and Chong (2002) note that prophylactic hyperventilation is associated with a poorer outcome; so traditional chronic hyperventilation should, therefore, be avoided. Transient episodes of hyperventilation, however, can be a rapid and useful method to limit the duration of acute and damaging increases in ICP (Iacono 2000; Mayer and Chong 2002; Lettieri 2006; BTF 2007).
Customary beliefs that the use of positive end-expiratory pressure (PEEP) will provoke a rise in ICP is no longer appropriate as hypoxaemia often fails to correct without the addition of PEEP and, with an adequate intravascular volume, a PEEP of 10 cmH2O does not elevate ICP and may even decrease it due to improved cerebral oxygenation (Huynh et al. 2002).
To maintain Paul’s endotracheal tube patency, mobilisation and removal of lung secretions is required. Unfortunately, chest physiotherapy and tracheal stimulation during suctioning could cause Paul adverse cerebrovascular and systemic vascular effects and may elevate his ICP (Yanko and Mitcho 2001). Endotracheal suctioning should be performed based on a Paul’s clinical signs and where benefits are considered to outweigh risks. Pre-oxygenation with FiO2 1.0 and minimising the number of catheter passes may avoid the cumulative effects that raise ICP (Yanko and Mitcho 2001).
Patient positioning is important and the head of the bed should be elevated to 30 degrees with Paul’s head and neck maintained in a straight and forward (neutral) alignment (Lee 1989; Feldman et al. 1992; Simmons 1997; Patterson et al. 2005). This position should reduce cerebral and jugular venous pressure, and promote venous drainage, without severely compromising CPP, CBF or cardiac output and may help protect against ventilator acquired pneumonia (Mayer and Chong 2002; Patterson et al. 2005; DH 2007).
Extreme flexion of Paul’s hips and knees should be avoided as it increases intra-abdominal pressure and hence intrathoracic pressure, which compromises venous drainage, increasing ICP (Yanko and Mitcho 2001). When turning Paul, a ‘log roll’ technique allows the head and neck to be kept in neutral alignment (Yanko and Mitcho 2001) (see Figures 12.2 and 12.3). If, as a precaution, Paul’s cervical spine is immobilised with a rigid collar, this can elevate ICP by approximately 4–6 mmHg due to compression of the jugular vein (Ho et al. 2002; Arbour 2004). The nurse should also be mindful when tying/securing Paul’s endotracheal tube tapes that jugular venous drainage is not impeded (Yanko and Mitcho 2001).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


