Targeted Therapies to Treat Cancer
Objectives
Key Terms
apoptosis, p. 533
cyclins, p. 532
cytotoxic, p. 530
mitosis, p. 531
monoclonal antibody, p. 547
multikinase inhibitors, p. 540
proteasome, p. 546
signal transduction, p. 531
suppressor gene, p. 532
targeted therapy, p. 531
telomeric DNA, p. 533
transcription factors, p. 532
tyrosine kinase inhibitor, p. 534
tyrosine kinase, p. 531
The authors gratefully acknowledge the work of Carolee A. Polek, who updated this chapter for the eighth edition.

As described in Chapter 37 and in the Unit XII opener, cancers develop from normal cells that have sustained gene damage. Cancer cells differ from normal cells in many ways, especially in their unrelenting growth and invasive spread (metastasis). This excessive growth serves no useful function and causes death when cancer cells invade normal tissues to the extent that vital organs can no longer perform their life-sustaining functions.
Traditional chemotherapy is generalized, systemic, chemical, cytotoxic treatment that directly kills or severely damages cells. Mechanisms of action for different categories of chemotherapy drugs vary, but the overall outcome is the inhibition of cell division (mitosis) and cell death. Often, these drugs damage the cell DNA to prevent DNA replication and formation of new cancer cells (see Chapter 37). This form of treatment has improved cancer control and increased long-term survival. Although these drugs have some selectivity for exerting cytotoxic effects on cancer cells, the drugs also have a toxic impact on many normal cells. As a result, acute side effects of traditional combination chemotherapy are uncomfortable and can be life-threatening. Thus, the cancer cell–killing effect of traditional chemotherapy is limited by the dosages and scheduling regimens needed to reduce toxic side effects on normal cells.
Targeted therapy for cancer treatment differs from traditional cancer chemotherapy by taking advantage of biologic features, such as cellular receptors, enzymes, pathways, or other molecular proteins of cancer cells that either are not present or are present in much smaller quantities in normal cells. Thus, targeted therapies are more specific in their mechanisms and effects than traditional cancer chemotherapy agents. The National Cancer Institute’s (NCI) defines targeted therapy as drugs or other substances that block the growth and spread of cancer by interfering with specific molecules involved in tumor growth and progression. It is important to remember that, unlike traditional cytotoxic chemotherapy (which exerts its effects by damaging the DNA of nearly any cell), targeted therapies require a specific molecular target as the recipient of their effects. The increased specificity of targeted therapies also means that more tests are required on cancer cells to determine whether a target is present in sufficient amounts to make the targeted therapy effective. For example, not all breast cancer cells have the molecular target for trastuzumab (Herceptin), and not all leukemia cells have the molecular target for imatinib (Gleevec). Cancers that do not have sufficient quantities of the specific molecular target will not respond to targeted therapy.
Pathophysiology
Normal Cell Growth Regulation
Genetic Control over Cell Division
As shown in Figure XII-1 at the beginning of Unit XII, which depicts the cell cycle, cell cycle checkpoints must be overcome to enter the cell cycle and continue to progress through each phase of the cycle to mitosis. Initially, general factors determine whether the cell enters the cell cycle.
Signal transduction is a method of communication that allows events, conditions, and substances outside of the cell to influence the cell’s decision to divide, not to divide, or to perform its designated function. Some of the known external and internal signaling substances that promote cell division are enzymes; growth factors; adhesion proteins; steroid hormones; and cell-to-cell physical, chemical, and electrical interactions. Figure 38-1 shows a segment of a cell with one interconnecting signal transduction pathway that, when activated, leads to gene activation for those proteins that promote cell division. This pathway can be activated by growth factors binding to their receptors, the interaction of certain drugs with the cell plasma membrane, the presence of adhesion proteins, changes in ion movement (especially sodium and calcium), ligand binding, and other cell-to-cell interactions. When any of these conditions activate the signal transduction pathway, the amount of tyrosine kinases inside the cell is increased.
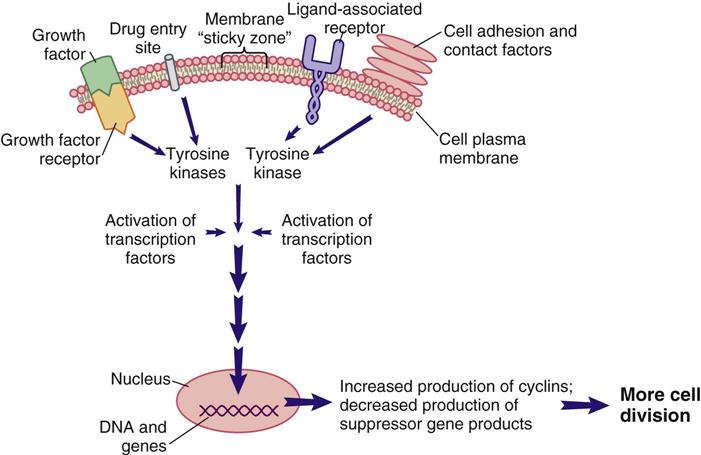
Tyrosine kinases (TKs) are a family of enzymes that activate other substances by adding a phosphate group (PO4) to them, a process known as phosphorylation. There are many different TKs. Some are unique to the cell type; others may be present only in cancer cells that express a specific gene mutation. Regardless of how a pro–cell-division signal transduction pathway is activated, the result is an increase in TK levels that propagate the signal by activating a variety of transcription factors within the pathway.
Transcription factors for cell division are substances that enter the nucleus and signal the cell that mitosis is needed. Many different types of substances serve as transcription factors. The overall response is greater expression of oncogene products (cyclins) that promote cell division, and the reduced expression of suppressor gene products that inhibit cell division. When the cell responds to these mitotic signals indicating that cell division is needed and resources are adequate, the cell leaves the G0 phase and enters the G1 phase of the cell cycle. Once the cell enters the G1 phase, the presence of specific cyclins determines whether it can progress to the next phase (Figure 38-2).
 Safety
Safety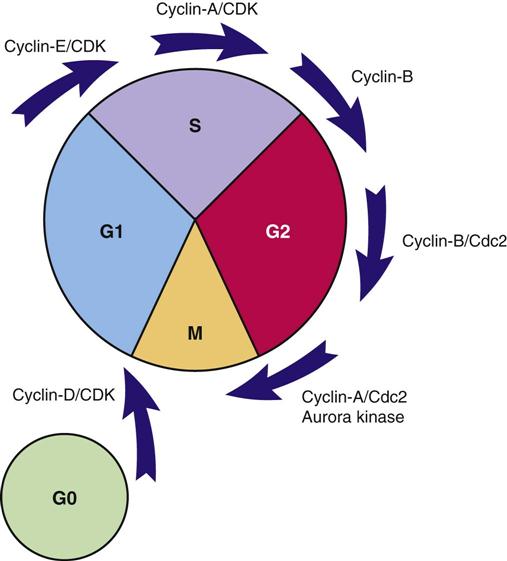
Cyclins are part of a family of proteins that, when active, stimulate the cell to move through the cell cycle. They are the products of oncogenes, and most are activated when a phosphorous group is added to the cyclin chemical structure. (Removal of a phosphorous molecule from a cyclin [dephosphorylation] inhibits its activity.) Kinases that activate cyclins are cyclin-dependent kinases (CDKs). CDKs combine with cyclins to form complexes that initiate cell mechanisms to complete cell division. In normal cells, the oncogenes that produce cyclins are carefully regulated by suppressor gene products so that cell division only occurs when it is needed and to the degree that it is needed.
The amount and type of cyclins as well as which specific CDK is present in the cell during cell division vary by the phase of the cycle. These differences in types of cyclins and CDKs determine when or if a particular cell moves from one phase of the cycle to the next. Many different groups of cyclins have been identified, with the D group being most well understood.
A common signal for entering and starting the cell cycle at G1 is the combining of a cyclin-D with the appropriate CDK to form a cyclin-D/CDK complex (see Figure 38-2). Movement of the cell from G1 into the S, G2, and M phases of the cell cycle is regulated by the continued presence of pro–cell-division transcription factors that promote DNA transcription and increased synthesis of specific pro–cell-division cyclins and CDKs.
Even when cell division is needed, the process is well controlled in normal cells. Proteins synthesized by suppressor genes determine how much oncogene expression is needed to allow cell division to occur but not lead to excessive cell division. Such control is exemplified by normal wound repair. For example, when a person falls and scrapes skin from the knee, the skin cells at the edge of the wound are signaled to divide and fill in the gap. When the wound area is closed, cell division normally stops. The person does not have uncontrolled cell division in this area to the extent that a large skin flap develops over and past the wound site.
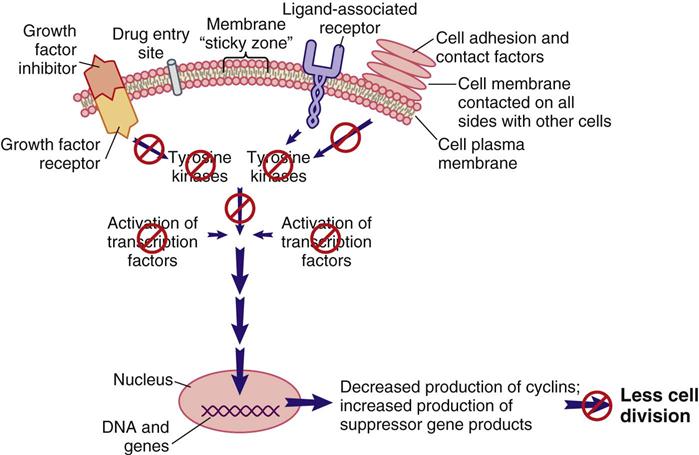
When cell division is not needed, external signals, such as growth factor inhibitors and the surrounding of a cell plasma membrane with other cells, send signals that are inhibitory to the pro–cell-division signal transduction pathway (Figure 38-3). The result of this inhibition leads to low levels of TKs and reduced levels of pro–cell-division transcription factors. Instead, suppressor gene activity is increased, resulting in production of more suppressor gene products that inhibit the synthesis of cyclins and CDKs by oncogenes. There are many suppressor genes, and, although all are present in every cell type, specific suppressor genes may be more active in selected types of tissues. For example, the BRCA1 suppressor gene appears most active in suppressing excessive cell division in breast, ovary, and genitourinary tract tissues. One of the most well-characterized suppressor genes is the Tp53 suppressor gene. Its gene product restricts the entry of cells into the cell cycle and restricts progression through the cell cycle for many cell types. Without suppressor gene products, oncogenes would be overexpressed continually, leading to uncontrolled and unneeded cell division.
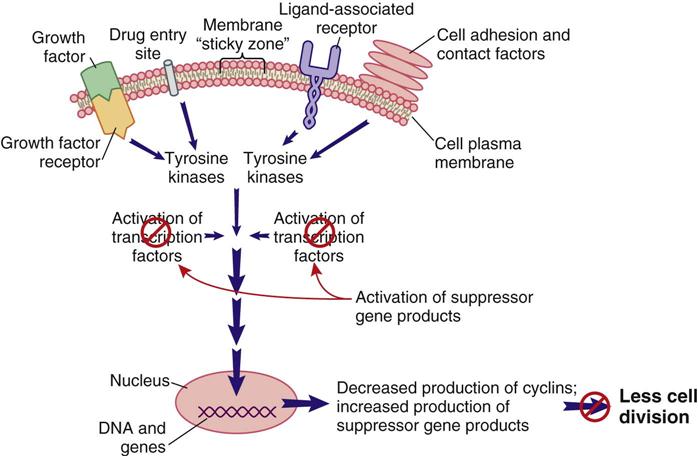
Internal cell conditions, such as poor cell nutrition and reduced energy stores, can trigger the activation of suppressor genes to disrupt the pro–cell-division signal transduction pathway, even when external conditions indicate a need for cell division (Figure 38-4). Thus, healthy and active suppressor genes guard against cell division when it is not in the body’s best interest.
Genetic Control over Cell Death
As some cells age, they begin to function less optimally. When a cell is damaged, reduced function occurs at an earlier cell age. In normal tissues that are capable of cell division, damaged cells and older cells respond to signals for apoptosis, which is programmed cell death, intended to ensure that tissues and organs contain only healthy and optimally functional cells. Apoptosis is under strict genetic control in normal tissues so that healthy functional cells do not self-destruct faster than they can be replaced, and that older or damaged cells unable to perform vital functions do not overpopulate a tissue and reduce organ efficiency. Maintenance of optimally functional organs depends on a balance of cell division with apoptosis.
The signals for apoptosis may come from the aging cell with the loss of telomeric DNA. Telomeric DNA is special DNA that caps the ends of each chromosome much like plastic tips cap the ends of a shoelace to prevent raveling (Figure 38-5). The function of telomeric DNA is to maintain the integrity of the double DNA strands within each chromosome. With each round of cell division, the telomeric DNA shortens. When the cell has undergone its lifespan’s worth of cell divisions, the telomeric DNA that capped the chromosomes is gone, allowing the DNA to unravel and fragment. These processes then trigger genetic and other intracellular signals for self-destruction through the action of autoenzymes, especially caspase 9. Activated caspase 9 leads to a cascade reaction for rapid activation of many more types of caspase enzymes. These enzymes degrade the cell’s internal structures and cause the plasma membrane to lyse and break the cell into small fragments that are removed from the body by white blood cells.
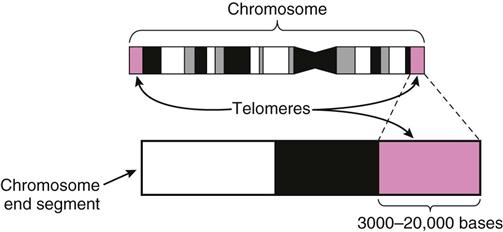
Apoptosis is regulated by different gene products, including those of the Tp53 (tumor protein 53) suppressor gene. When cells reach a certain age or experience DNA damage, the Tp53 gene is expressed and older or damaged cells either undergo apoptosis or are prohibited from progression through the cell cycle.
Growth Regulation and Cancer
Loss of Genetic Control of Cell Growth.
As stated earlier in the chapter, cancers develop from cells that were once normal. Normal cells become cancer cells when external or internal conditions lead to gene damage. Although cancer can develop from a normal cell that has sustained damage to its oncogenes, most commonly, one or more suppressor genes are damaged and are no longer able to control oncogene expression. As a result of excessive oncogene expression, cyclins and CDKs are overproduced and cell division occurs when it is not needed.
Excessive cell division from gene damage/mutations appears to be self-perpetuating, leading to further gene mutations that do the following:
Loss of Apoptosis.
Mutations that occur in suppressor genes as a result of DNA damage inactivate the suppressor genes, preventing them from controlling oncogene activity. Some suppressor genes, such as the Tp53 gene, also regulate apoptosis. Inactivation of suppressor genes that regulate apoptosis makes cancer cells unresponsive to apoptotic signals. These cancer cells are now resistant to natural cell death, a feature known as cellular immortality. The combined effects of lack of regulation for cell division and the loss of apoptosis result in cancer cells having no balance between cell division and apoptosis. This unbalanced condition favors continuous cancer cell division.
Targeted Therapy Drugs
Targeted cancer therapies are a relatively new approach in cancer treatment. They work by interfering with cancer cell growth and division in different ways. These drugs are broadly known as signal transduction inhibitors. Targeted therapies generally inhibit cancer cell division by blocking a cancer membrane receptor, blocking tyrosine kinase activity, interfering with signal transduction, stimulating an immune system attack on cancer cells, or inducing the cell to undergo apoptosis. These agents may be used as monotherapy (a single agent), in combination with traditional chemotherapy, and with radiotherapy. Some targeted therapies are oral and are self-administered at home where it is convenient for patients; others are administered parenterally. All targeted therapies to treat cancer are categorized as high-alert medications. 
The rapid identification of specific cancer-cell targets in recent years has led to an increased development of targeted therapies. Some are in relatively common use, and others are used less frequently or for rarer cancer types. Management of patient issues related to targeted therapies is an evolving area of study. With many targeted therapies being new to the market, costs can be high and may not be covered by insurance. New targeted therapies are approved frequently. Older targeted therapy drugs may be newly approved for use in a different way or with a different cancer type. Expect to see indications for use of different targeted therapies in additional cancer types in the future. Because newly approved therapies have been less widely used and their adverse effects are not yet fully characterized, this chapter focuses on therapies that have been in use for 1 year or more.
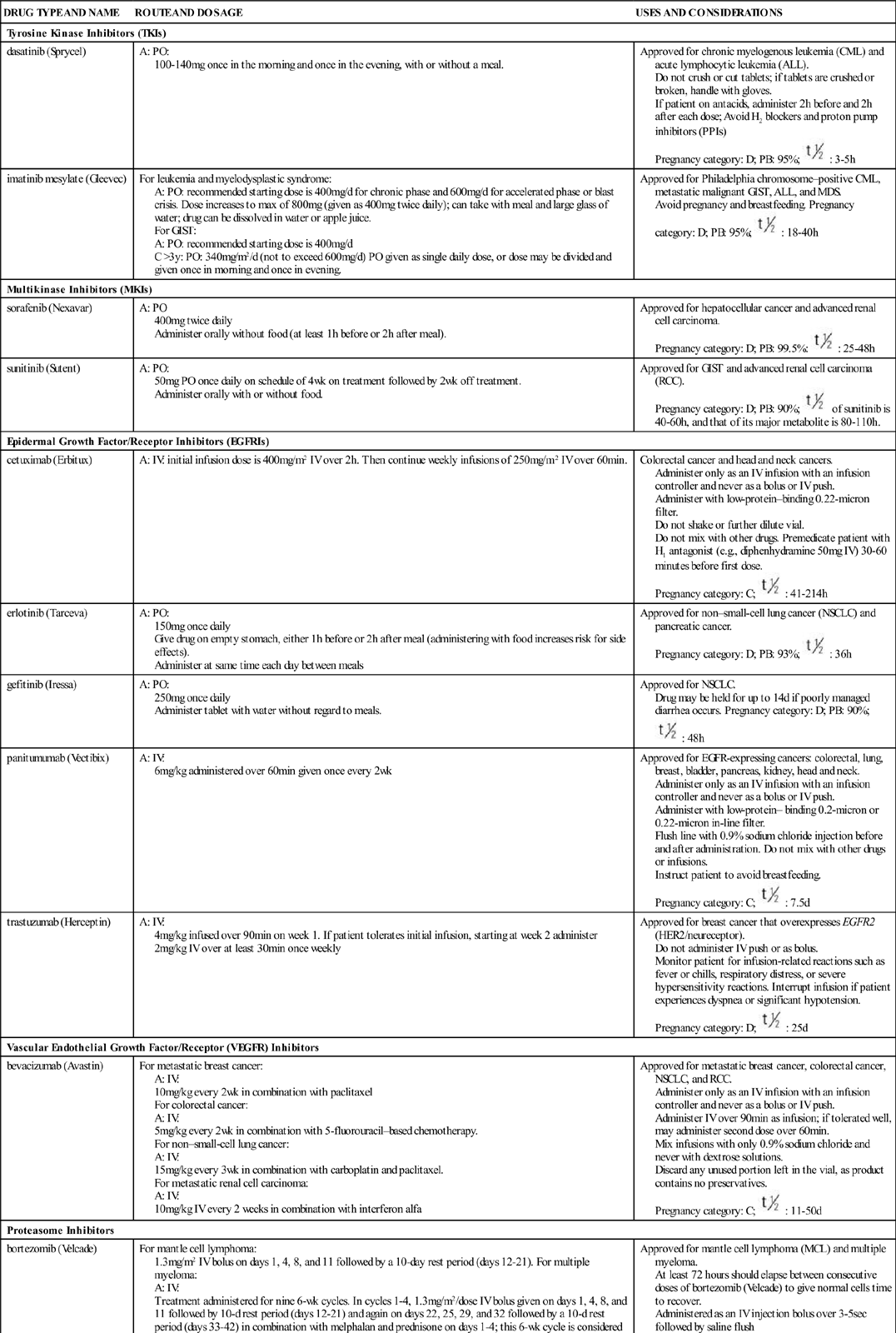
There are several major classes of targeted cancer therapies, based on their most common mechanism of action (Table 38-1). Some targeted therapies have more than one action. Those discussed in this chapter include tyrosine kinases inhibitors, multikinase inhibitors, epidermal growth factor/receptor inhibitors, vascular endothelial growth factor/receptor inhibitors, proteasome inhibitors, angiogenesis inhibitors, and monoclonal antibodies. The targeted therapies have been divided into two categories: monoclonal antibody (MoAb) and small-molecule inhibitor.
TABLE 38-1
 TARGETED THERAPIES FOR CANCER TREATMENT
TARGETED THERAPIES FOR CANCER TREATMENT
| DRUG TYPE AND NAME | ROUTE AND DOSAGE | USES AND CONSIDERATIONS |
| Tyrosine Kinase Inhibitors (TKIs) | ||
| dasatinib (Sprycel) | A: PO: 100-140 mg once in the morning and once in the evening, with or without a meal. | Approved for chronic myelogenous leukemia (CML) and acute lymphocytic leukemia (ALL). Do not crush or cut tablets; if tablets are crushed or broken, handle with gloves. If patient on antacids, administer 2 h before and 2 h after each dose; Avoid H2 blockers and proton pump inhibitors (PPIs) Pregnancy category: D; PB: 95%; 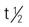 : 3-5 h : 3-5 h |
| imatinib mesylate (Gleevec) | For leukemia and myelodysplastic syndrome: A: PO: recommended starting dose is 400 mg/d for chronic phase and 600 mg/d for accelerated phase or blast crisis. Dose increases to max of 800 mg (given as 400 mg twice daily); can take with meal and large glass of water; drug can be dissolved in water or apple juice. For GIST: A: PO: recommended starting dose is 400 mg/d C >3 y: PO: 340 mg/m2/d (not to exceed 600 mg/d) PO given as single daily dose, or dose may be divided and given once in morning and once in evening. | Approved for Philadelphia chromosome–positive CML, metastatic malignant GIST, ALL, and MDS. Avoid pregnancy and breastfeeding. Pregnancy category: D; PB: 95%; 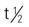 : 18-40 h : 18-40 h |
| Multikinase Inhibitors (MKIs) | ||
| sorafenib (Nexavar) | A: PO 400 mg twice daily Administer orally without food (at least 1 h before or 2 h after meal). | Approved for hepatocellular cancer and advanced renal cell carcinoma. Pregnancy category: D; PB: 99.5%: 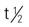 : 25-48 h : 25-48 h |
| sunitinib (Sutent) | A: PO: 50 mg PO once daily on schedule of 4 wk on treatment followed by 2 wk off treatment. Administer orally with or without food. | Approved for GIST and advanced renal cell carcinoma (RCC). Pregnancy category: D; PB: 90%; 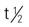 of sunitinib is 40-60 h, and that of its major metabolite is 80-110 h. of sunitinib is 40-60 h, and that of its major metabolite is 80-110 h. |
| Epidermal Growth Factor/Receptor Inhibitors (EGFRIs) | ||
| cetuximab (Erbitux) | A: IV: initial infusion dose is 400 mg/m2 IV over 2 h. Then continue weekly infusions of 250 mg/m2 IV over 60 min. | Colorectal cancer and head and neck cancers. Administer only as an IV infusion with an infusion controller and never as a bolus or IV push. Administer with low-protein–binding 0.22-micron filter. Do not shake or further dilute vial. Do not mix with other drugs. Premedicate patient with H1 antagonist (e.g., diphenhydramine 50 mg IV) 30-60 minutes before first dose. Pregnancy category: C; 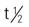 : 41-214 h : 41-214 h |
| erlotinib (Tarceva) | A: PO: 150 mg once daily Give drug on empty stomach, either 1 h before or 2 h after meal (administering with food increases risk for side effects). Administer at same time each day between meals | Approved for non–small-cell lung cancer (NSCLC) and pancreatic cancer. Pregnancy category: D; PB: 93%; 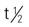 : 36 h : 36 h |
| gefitinib (Iressa) | A: PO: 250 mg once daily Administer tablet with water without regard to meals. | Approved for NSCLC. Drug may be held for up to 14 d if poorly managed diarrhea occurs. Pregnancy category: D; PB: 90%; 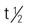 : 48 h : 48 h |
| panitumumab (Vectibix) | A: IV: 6 mg/kg administered over 60 min given once every 2 wk | Approved for EGFR-expressing cancers: colorectal, lung, breast, bladder, pancreas, kidney, head and neck. Administer only as an IV infusion with an infusion controller and never as a bolus or IV push. Administer with low-protein– binding 0.2-micron or 0.22-micron in-line filter. Flush line with 0.9% sodium chloride injection before and after administration. Do not mix with other drugs or infusions. Instruct patient to avoid breastfeeding. Pregnancy category: C; 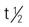 : 7.5 d : 7.5 d |
| trastuzumab (Herceptin) | A: IV: 4 mg/kg infused over 90 min on week 1. If patient tolerates initial infusion, starting at week 2 administer 2 mg/kg IV over at least 30 min once weekly | Approved for breast cancer that overexpresses EGFR2 (HER2/neureceptor). Do not administer IV push or as bolus. Monitor patient for infusion-related reactions such as fever or chills, respiratory distress, or severe hypersensitivity reactions. Interrupt infusion if patient experiences dyspnea or significant hypotension. Pregnancy category: D; 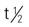 : 25 d : 25 d |
| Vascular Endothelial Growth Factor/Receptor (VEGFR) Inhibitors | ||
| bevacizumab (Avastin) | For metastatic breast cancer: A: IV: 10 mg/kg every 2 wk in combination with paclitaxel For colorectal cancer: A: IV: 5 mg/kg every 2 wk in combination with 5-fluorouracil–based chemotherapy. For non–small-cell lung cancer: A: IV: 15 mg/kg every 3 wk in combination with carboplatin and paclitaxel. For metastatic renal cell carcinoma: A: IV: 10 mg/kg IV every 2 weeks in combination with interferon alfa | Approved for metastatic breast cancer, colorectal cancer, NSCLC, and RCC. Administer only as an IV infusion with an infusion controller and never as a bolus or IV push. Administer IV over 90 min as infusion; if tolerated well, may administer second dose over 60 min. Mix infusions with only 0.9% sodium chloride and never with dextrose solutions. Discard any unused portion left in the vial, as product contains no preservatives. Pregnancy category: C; 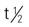 : 11-50 d : 11-50 d |
| Proteasome Inhibitors | ||
| bortezomib (Velcade) | For mantle cell lymphoma: 1.3 mg/m2 IV bolus on days 1, 4, 8, and 11 followed by a 10-day rest period (days 12-21). For multiple myeloma: A: IV: Treatment administered for nine 6-wk cycles. In cycles 1-4, 1.3 mg/m2/dose IV bolus given on days 1, 4, 8, and 11 followed by 10-d rest period (days 12-21) and again on days 22, 25, 29, and 32 followed by a 10-d rest period (days 33-42) in combination with melphalan and prednisone on days 1-4; this 6-wk cycle is considered one course. | Approved for mantle cell lymphoma (MCL) and multiple myeloma. At least 72 hours should elapse between consecutive doses of bortezomib (Velcade) to give normal cells time to recover. Administered as an IV injection bolus over 3-5 sec followed by saline flush Reconstitute for injection each vial with 3.5 mL of 0.9% sodium chloride for a final concentration of 1 mg/mL. Final product should be a clear, colorless, solution. Pregnancy category: D; PB: 83%: 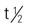 : 9-15 h : 9-15 h |
| Angiogenesis Inhibitors | ||
| temsirolimus (Torisel) | A: IV: 25 mg IV given over 30-60 min once a wk | Approved for advanced RCC. During preparation, protect drug from excessive room light and sunlight. Premedicate with diphenhydramine (Benadryl) 25-50 mg IV, or a similar antihistamine, approximately 30 min before the start of each dose. Two dilutions are required before IV infusion. Use only supplied diluent for initial dilution. Use an in-line polyethersulfone filter with pore size of ≤5 microns and an infusion pump. Administer only through polyethylene-lined administration sets. Administer dose over 30-60 min. Pregnancy category: D;  : 17-20 d : 17-20 d |
| Monoclonal Antibodies | ||
| alemtuzumab (Campath) | A: IV: Start at 3 mg IV over 2 h once daily. When 3-mg dose is tolerated (infusion reactions grade 2 or less), escalate daily dose to 10 mg IV once daily and continue until infusion reactions are grade 2 or less, then start the maintenance dose of 30 mg IV given 3 times weekly on alternate days (e.g., Monday, Wednesday, Friday); total therapy duration including dose escalation is 12 wk. | Approved for chronic lymphocytic leukemia (CLL). Administer as an IV infusion with an infusion controller over 2 h and never as a bolus or IV push. Monitor patient closely for serious, sometimes fatal, infusion-related reactions. Premedicate 30 min before first dose, with any dose escalation, and as needed with diphenhydramine (Benadryl) (50 mg) and acetaminophen (Tylenol) (500-1000 mg). Withhold alemtuzumab (Campath) administration if a grade 3 or 4 infusion reaction occurs Pregnancy category: C;  : 12 d : 12 d |
| ibritumomab tiuxetan (Zevalin) | A: IV: Step 1: Rituximab 250 mg/m2 IV is administered first (see rituximab monograph). Within 4 h of completing rituximab infusion, give 5 mCi (1.6 mg total antibody dose) of In-111 ibritumomab tiuxetan IV over 10 min. Step 2: Seven to nine d following step 1, give a second dose of rituximab 250 mg/m2 IV. Within 4 h of completing the rituximab infusion, give Y-90 ibritumomab tiuxetan 0.4 mCi/kg (14.8 MBq/kg) or 0.3 mCi/kg (11.1 MBq/kg) IV over 10 min. | Approved for B-cell NHL. Use appropriate precautions for handling, preparing, and administering radiopharmaceuticals. Do not administer In-111 ibritumomab tiuxetan (Zevalin) and Y-90 ibritumomab tiuxetan unless rituximab (Rituxan) predose has been administered. Do not give Y-90 ibritumomab tiuxetan if platelet count <100,000/mm3). Pregnancy category: D;  : 30 h : 30 h |
| rituximab (Rituxan) | A: IV: 375 mg/m2 once weekly for 4 doses; may be retreated for additional 4 doses. | Relapsed or refractory, low-grade, or follicular CD20-positive B-cell NHL. Premedicate 30 min before administration with diphenhydramine (Benadryl) (50 mg), acetaminophen (Tylenol) 650-1000 mg (adults), and possibly corticosteroids. Do not administer as IV push or bolus. Administer first IV infusion at initial rate of 50 mg/h. If no hypersensitivity or infusion-related events, increase infusion rate in 50 mg/h increments every 30 min, to a maximum of 400 mg/h. Monitor patients closely during infusion for profound hypotension, dyspnea, and cardiovascular infusion–related reactions. Do not mix rituximab solution with other drugs. Pregnancy category: C;  : 31.5-52.6 h : 31.5-52.6 h |
| I-131 tositumomab (Bexxar) (radioimmunotherapeutic agent) | A: IV: Step 1: Dosimetric step: On day 0, give 450 mg unlabeled tositumomab IV over 1 h followed by I-131 tositumomab 5 mCi (35 mg tositumomab) IV over 20 min. Total body gamma counts using a gamma camera obtained on day 0; day 2, 3, or 4; and day 6 or 7. Using these counts, perform calculations based on standard internal radiation dosimetry methods to determine patient-specific activity (in mCi) of radiolabeled tositumomab required to deliver maximum tolerated dose of 75 cGy total-body dose. Step 2: Therapeutic step: 450 mg unlabeled tositumomab IV over 1 h followed by patient-specific activity (in millicuries) iodine I-131 labeled to 35 mg of tositumomab IV between days 7 and 14. | Refractory CD20-positive, follicular NHL. Administer thyroid protection agents at least 24 h before step 1 and step 2. Do not administer to patients with known murine (mouse) hypersensitivity. Premedicate 30 min before administration with oral diphenhydramine (Benadryl) (50 mg) and acetaminophen (Tylenol) 650-1000 mg (adults). Use appropriate precautions for handling, preparing, and administering radiopharmaceuticals. Administer through IV tubing set with 0.22-micron in-line filter. Use same IV tubing set and filter throughout entire dosimetric or therapeutic step. Administer iodine I-131 tositumomab (Bexxar) infusion over 60 min. After infusion of iodine I-131 tositumomab (Bexxar) is complete, close stopcock to syringe. Flush extension set and secondary IV infusion set with 0.9% sodium chloride for injection. Pregnancy category: X; 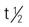 : 8.5 d : 8.5 d |
Tyrosine Kinase Inhibitors
Several types of targeted therapies have as an outcome the inhibition of tyrosine kinases using a variety of mechanisms. Specific drugs that cause this action as their main mechanisms are referred to as tyrosine kinase inhibitors (TKIs). The most common TKIs currently prescribed are imatinib mesylate (Gleevec) and dasatinib (Sprycel). Table 38-1 lists the TKIs and their dosages, routes, uses, and considerations. Prototype Drug Chart 38-1 lists specific drug information about the TKI imatinib (Gleevec). Other approved TKIs include lapatinib (Tykerb), which is used with other drugs in the treatment of HER2-positive breast cancer. 
 Prototype Drug Chart 38-1
Prototype Drug Chart 38-1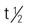 of drug is 18 hours, of its major metabolite 40 hours.
of drug is 18 hours, of its major metabolite 40 hours.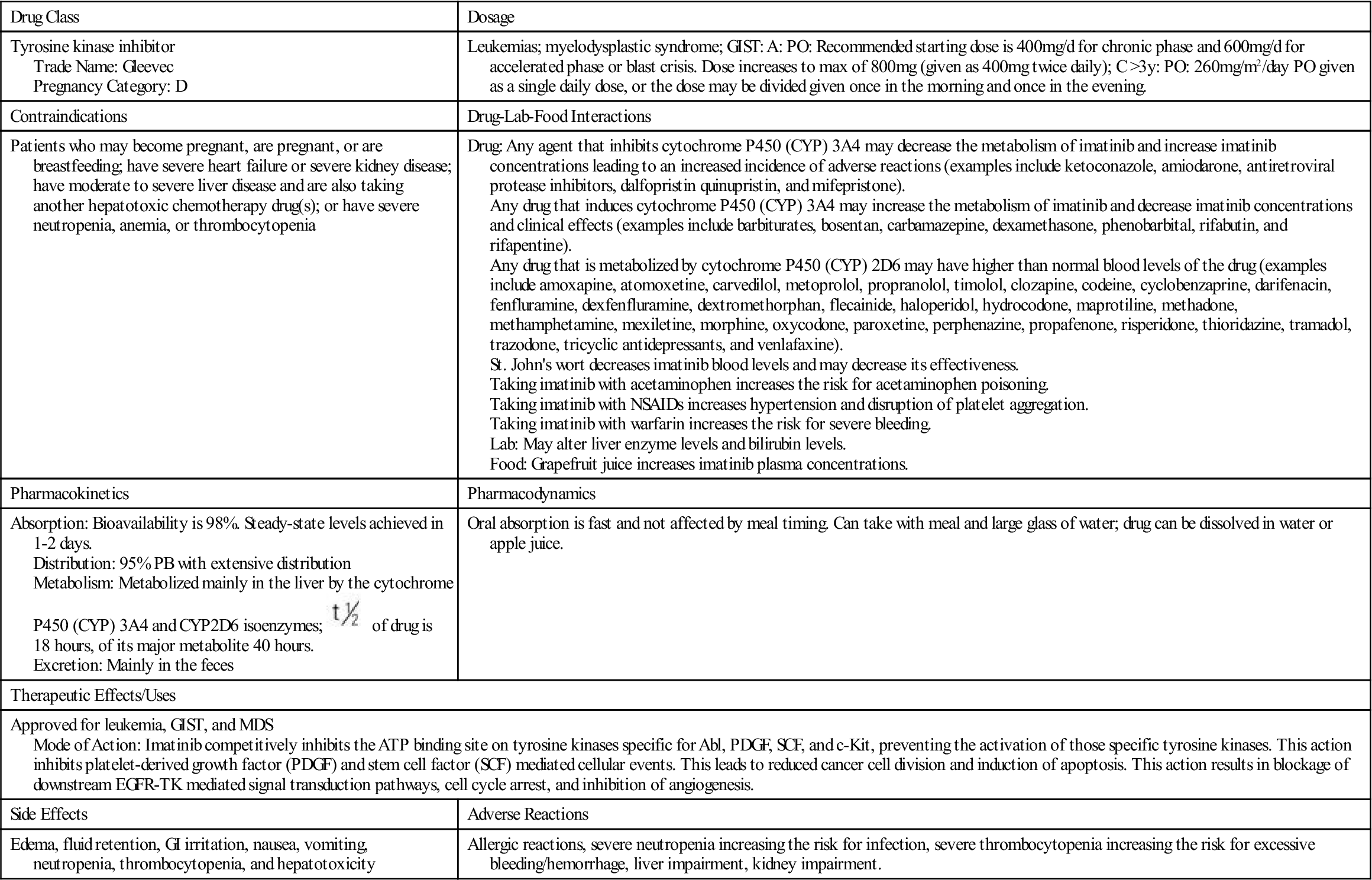
 , half-life; y, year; >, greater than.
, half-life; y, year; >, greater than.TKIs exert their effects by directly inhibiting only specific types of tyrosine kinases. The types they inhibit are the SRC kinases, which are present in many cells (normal and cancerous) and a very specific type, the BCR-ABL tyrosine kinase. As shown in Figure 38-1, receptors on the cell membrane can activate tyrosine kinases, which then turn on signal transduction pathways promoting cell division. The TKIs are nonreceptor kinase inhibitors because they do not bind to the receptors on the plasma membrane. Instead they work directly on the tyrosine kinase molecule.
The SRC kinases are involved in the activation of signal transduction pathways promoting cell division in many cell types. The BCR-ABL tyrosine kinase is present only in cancer cells that have a specific gene mutation resulting from a chromosome structural rearrangement that forms a “Philadelphia chromosome.” This mutation is highly present in chronic myelogenous leukemia (CML) cells and has been found in some other cancer cell types. When activated and expressed, BCR-ABL tyrosine kinase turns on a strong pro–cell-division signal transduction pathway that leads to proliferation of cancer cells. TKIs prevent activation of tyrosine kinases, which then inhibits further activation of the signal transduction pathway and stops the proliferation of cancer cells. This action can control the disease but cannot alone eradicate it.
Dasatinib
Dasatinib (Sprycel) is approved for chronic myelogenous leukemia (CML) and acute lymphocytic leukemia (ALL).
Pharmacokinetics
Dasatinib is an oral drug that is readily absorbed from the gastrointestinal (GI) tract and can be taken with or without food. Antacids slow the absorption. The drug is extensively metabolized in the liver by the cytochrome P450 (CYP) 3A4 isoenzyme.
Pharmacodynamics
Dasatinib most specifically targets BCR-ABL tyrosine kinase, found in CML cells. The drug blocks the ATP (adenosine triphosphase) binding site on the tyrosine kinase so that it does not become activated. This lack of BCR-ABL tyrosine kinase activation inhibits further activation of the signal transduction pathway and stops the proliferation of cancer cells. The drug has the greatest effects on cancer cells expressing the Philadelphia chromosome genetic mutation.
Side Effects and Adverse Reactions.
Dasatinib often causes electrolyte imbalances, especially low serum levels of phosphorus and calcium. These imbalances may require phosphorus and calcium replacement. ECG abnormalities, especially prolonged QT interval, have been seen in patients taking dasatinib. This drug should be used cautiously in anyone at risk for long QT, such as those who have hypokalemia or hypomagnesemia and those with a family history of long QT syndrome. Fluid retention has also been seen in up to 50% of patients. Hematologic side effects of myelosuppression with anemia, thrombocytopenia, and neutropenia are relatively common.
Drug Interactions.
CYP3A4 enzyme inhibitors decrease dasatinib metabolism, resulting in an increased serum concentration and increased risk for toxicity. Other drugs that strongly increase the serum concentration of dasatinib include atazanavir, clarithromycin, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, triazolobenzodiazepines, and voriconazole.
Drugs that enhance the activity of the CYP3A4 enzyme decrease dasatinib serum levels and reduce its effectiveness. Such drugs include aminoglutethimide, barbiturates, carbamazepine, dexamethasone, griseofulvin, modafinil, nafcillin, phenytoin, primidone, rifabutin, and rifampin. Other drugs that appear to reduce the effectiveness of dasatinib include antacids, H2 blockers, and proton pump inhibitors.
Dasatinib may interfere with the metabolism of other drugs that use the CYP3A4 enzyme system, such as alfentanil, astemizole, terfenadine, cisapride, cyclosporine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus, and ergot alkaloids.
Multikinase Inhibitors
The multikinase inhibitors (MKIs) are chemicals that directly inhibit the activity of specific kinases in cancer cells and in cancer cell vasculature. (Recall that kinases are enzymes that activate other proteins, including those that activate signal transduction pathways promoting cancer cell division.) Table 38-1 lists the MKIs and their dosages, routes, uses, and considerations.
Sorafenib
Sorafenib (Nexavar) is a multikinase inhibitor that specifically targets serine/threonine and receptor tyrosine kinases, which are activated as a result of gene mutations and are most commonly found in pancreatic cancer, colon cancer, and non–small-cell lung cancer. In addition, the drug may be used in those hepatocellular carcinomas and renal cell carcinomas that overexpress the target.
Pharmacokinetics
Sorafenib (Nexavar) is an oral drug whose absorption and bioavailability are inhibited when taken with a high-fat meal. The drug is metabolized in the liver, mainly by the CYP3A4 enzyme system. It is 99.5% protein-bound and reaches peak plasma level in about 3 hours. The drug is eliminated in the feces and urine, with a half-life of 25 to 48 hours.
Pharmacodynamics
Sorafenib inhibits Raf kinase, vascular endothelial growth factor (VEGF) receptors VEGFR-2 and VEGFR-3, platelet-derived growth factor receptor (PDGFR), Kit receptor tyrosine kinase (KIT), fms-like tyrosine kinase 3 (FLT-3), and RET. When cytokines or growth factors activate these tyrosine kinase receptors, a protein-kinase–mediated cascade starts, leading to uncontrolled cellular proliferation. The inhibition of these signaling pathways by sorafenib results in decreased cancer cellular proliferation. In addition, sorafenib specifically inhibits two VEGF receptors (VEGFR-2 and VEGFR-3), which are key receptor tyrosine kinases involved in angiogenesis. This action results in reduced blood vessel formation in cancer cells. This anticancer activity is present only when the drug is present, and cancer growth returns when therapy is completed.
Side Effects and Adverse Reactions.
Sorafenib has many common side effects. Hypertension is very common and can occur within the first 6 weeks of therapy. Skin side effects include alopecia, pruritus, dry skin, exfoliative dermatitis, acne, flushing, and palmar-plantar erythrodysesthesia (hand-foot syndrome), which also manifest within the first 6 weeks. Other side effects include weight loss, nausea/vomiting, diarrhea, anorexia, constipation, abdominal pain, mucositis, dyspepsia, dysphagia, and mild neutropenia and thrombocytopenia. More severe effects are possible, including pancreatitis, erectile dysfunction, and myocardial ischemia.
Drug Interactions.
Sorafenib levels are not increased by the presence of other drugs, even those that inhibit the enzyme that metabolizes sorefenib. However, drugs that induce metabolizing enzyme activity can reduce the blood levels and effectiveness of sorafenib. These include rifampin, phenytoin, phenobarbital, carbamazepine, dexamethasone, rifabutin, and rifapenten.
Sorafenib can increase the blood levels of rapaglinide, amiodarone, ibuprofen, loperamide, irinotecan, propofol, and warfarin.
Sunitinib
Sunitinib (Sutent) inhibits more than 80 tyrosine kinases. This inhibition results in regression of tumor growth, especially in clear cell renal cell carcinoma (RCC) and gastrointestinal stromal tumors (GIST).
Pharmacokinetics
Sunitinib is an oral drug that is well absorbed with or without meals. It is 90% protein-bound and reaches peak plasma levels in 6 to 12 hours. The drug is metabolized in the liver, mainly by the CYP3A4 enzyme system, and is eliminated in the feces. The half-life of sunitinib is 40 to 60 hours, and its major active metabolite has a half-life of 80 to 110 hours.
Pharmacodynamics
The action of sunitinib is the inhibition of many receptor tyrosine kinases (RTKs), including those of platelet-derived growth factor receptors (PDGFR), vascular endothelial growth factor receptors (VEGFR1, VEGFR2, VEGFR3), and a variety of others. This inhibition results in decreased cancer cell proliferation and in reduced blood vessel formation in cancer cells. This drug also increases adverse effects on normal tissues, especially hair and skin.
Side Effects and Adverse Reactions.
Side effects and adverse reactions are more widespread as a result of the number of different types of kinases sunitinib inhibits. Cardiovascular effects include hypertension, peripheral edema, left ventricular dysfunction, prolonged QT interval, and venous thromboembolism. GI effects include nausea/vomiting, diarrhea, stomatitis, and dyspepsia. Neuromuscular effects include fatigue, asthenia, headache, dizziness, peripheral neuropathy, mild arthralgia, limb pain, myalgia, and back pain. Liver impairment may occur with elevated liver enzyme levels and jaundice of the skin and sclera. Common integumentary changes include depigmentation of the skin, skin discoloration, rash, dry skin, and palmar-plantar erythrodysesthesia. Endocrine changes may include hypothyroidism and adrenal insufficiency. Hematologic changes may include myelosuppression. Respiratory-associated effects may include mild dyspnea and cough.
Drug Interactions.
Sunitinib’s blood levels and activity are increased by drugs that inhibit CYP3A4 enzyme levels, including atazanavir, clarithromycin, ketoconazole, itraconazole, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole, diltiazem, and verapamil. When these drugs are used during sunitinib therapy, side effects of sunitinib are more common and more severe. Drugs that increase sunitinib elimination and reduce its effectiveness include rifampin, phenytoin, phenobarbital, carbamazepine, dexamethasone, rifabutin, and rifapentin.
Epidermal Growth Factor/Receptor Inhibitors
The epidermal growth factor/receptor inhibitors (EGFRIs) include erlotinib, gefitinib, panitumumab, and cetuximab. Most EGFRIs ultimately also inhibit tyrosine kinase, but they do it more indirectly than the TKIs. As shown in Figure 38-1, the growth factor receptors on the cell membrane can activate tyrosine kinases, which then turn on signal transduction pathways promoting cell division. The EGFRIs bind to different areas of the epidermal growth factor receptor, blocking its activity so that it cannot activate tyrosine kinase. As a result, the downstream signal transduction pathway for promotion of cell division is inhibited and cell proliferation is severely limited. Table 38-1 lists the EGFRIs and their dosages, routes, uses, and considerations. Prototype Drug Chart 38-2 lists the specific drug information for erlotinib (Tarceva). 
 Nursing Process
Nursing Process






