Drug Action
Pharmaceutic, Pharmacokinetic, and Pharmacodynamic Phases
Objectives
• Differentiate the three phases of drug action.
• Discuss the two processes that occur before tablets are absorbed into the body.
• Describe the four processes of pharmacokinetics.
• Describe the nursing implications of pharmacokinetics and pharmacodynamics.
Key Terms
active absorption, p. 3
adverse reactions, p. 11
agonists, p. 8
antagonists, p. 8
bioavailability, p. 4
creatinine clearance, p. 7
disintegration, p. 3
dissolution, p. 3
distribution, p. 4
duration of action, p. 7
elimination, p. 7
excipients, p. 3
first-pass effect, p. 4
free drugs, p. 5
half-life, p. 6
ligand-binding domain, p. 8
loading dose, p. 10
metabolism, p. 6
nonselective drugs, p. 8
nonspecific drugs, p. 8
onset of action, p. 7
passive absorption, p. 3
peak action, p. 7
peak drug level, p. 10
pharmaceutic phase, p. 3
pharmacodynamics, p. 7
pharmacogenetics, p. 11
pharmacokinetics, p. 3
pinocytosis, p. 4
placebo effect, p. 11
protein-binding effect, p. 4
receptor families, p. 8
side effects, p. 10
tachyphylaxis, p. 11
therapeutic index, p. 9
therapeutic range (therapeutic window), p. 9
time-response curve, p. 7
tolerance, p. 11
toxic effects, p. 11
toxicity, p. 11
trough drug level, p. 10
 http://evolve.elsevier.com/KeeHayes/pharmacology/
http://evolve.elsevier.com/KeeHayes/pharmacology/
The authors gratefully acknowledge the work of Marilyn Herbert-Ashton, who updated this chapter for the eighth edition.
A tablet or capsule taken by mouth goes through three phases—pharmaceutic, pharmacokinetic, and pharmacodynamic—as drug actions occur. In the pharmaceutic phase, the drug becomes a solution so that it can cross the biologic membrane. When the drug is administered parenterally by subcutaneous (subQ), intramuscular (IM), or intravenous (IV) routes, there is no pharmaceutic phase. The second phase, the pharmacokinetic phase, is composed of four processes: absorption, distribution, metabolism (or biotransformation), and excretion (or elimination). In the pharmacodynamic phase, a biologic or physiologic response results.
Pharmaceutic Phase
Approximately 80% of drugs are taken by mouth. The pharmaceutic phase (dissolution) is the first phase of drug action. In the gastrointestinal (GI) tract, drugs need to be in solution so they can be absorbed. A drug in solid form (tablet or capsule) must disintegrate into small particles to dissolve into a liquid, a process known as dissolution. Drugs in liquid form are already in solution. Figure 1-1 displays the pharmaceutic phase of a tablet.

Tablets are not 100% drug. Fillers and inert substances, generally called excipients, are used in drug preparation to allow the drug to take on a particular size and shape and to enhance drug dissolution. Some additives in drugs, such as the ions potassium (K) and sodium (Na) in penicillin potassium and penicillin sodium, increase the absorbability of the drug. Penicillin is poorly absorbed by the GI tract because of gastric acid. However, by making the drug a potassium or sodium salt, penicillin can then be absorbed. Disintegration is the breakdown of a tablet into smaller particles. Dissolution is the dissolving of the smaller particles in the GI fluid before absorption. Rate of dissolution is the time it takes the drug to disintegrate and dissolve to become available for the body to absorb it. Drugs in liquid form are more rapidly available for GI absorption than are solids. Generally, drugs are both disintegrated and absorbed faster in acidic fluids with a pH of 1 or 2 rather than in alkaline fluids. Alkaline drugs would become ionized and have difficulty crossing cell membrane barriers. Both the very young and older adults have less gastric acidity; therefore, drug absorption is generally slower for those drugs absorbed primarily in the stomach.
Enteric-coated drugs resist disintegration in the gastric acid of the stomach, so disintegration does not occur until the drug reaches the alkaline environment of the small intestine. Enteric-coated tablets can remain in the stomach for a long time; therefore, their effect may be delayed in onset. Enteric-coated tablets or capsules and sustained-release (beaded) capsules should not be crushed. Crushing would alter the place and time of absorption of the drug. Food in the GI tract may interfere with the dissolution of certain drugs. Some drugs irritate the gastric mucosa, so fluids or food may be necessary to dilute the drug concentration and to act as protectants.
Pharmacokinetic Phase
Pharmacokinetics is the process of drug movement to achieve drug action. The four processes are absorption, distribution, metabolism (or biotransformation), and excretion (or elimination). The nurse applies knowledge of pharmacokinetics when assessing the patient for possible adverse drug effects. The nurse communicates assessment findings to members of the health care team in a timely manner to promote safe and effective drug therapy for the patient.
Absorption
Absorption is the movement of drug particles from the GI tract to body fluids by passive absorption, active absorption, or pinocytosis. Most oral drugs are absorbed into the surface area of the small intestine through the action of the extensive mucosal villi. Absorption is reduced if the villi are decreased in number because of disease, drug effect, or the removal of small intestine. Protein-based drugs such as insulin and growth hormones are destroyed in the small intestine by digestive enzymes. Passive absorption occurs mostly by diffusion (movement from higher concentration to lower concentration). With the process of diffusion, the drug does not require energy to move across the membrane. Active absorption requires a carrier such as an enzyme or protein to move the drug against a concentration gradient. Energy is required for active absorption. Pinocytosis is a process by which cells carry a drug across their membrane by engulfing the drug particles (Figure 1-2).
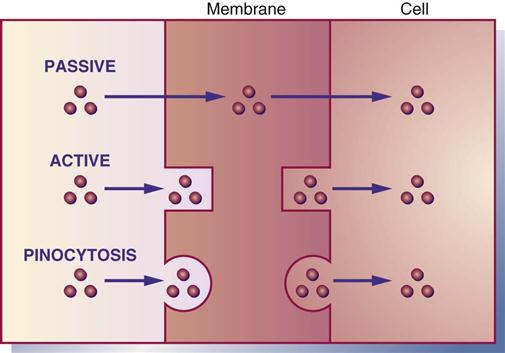
The GI membrane is composed mostly of lipid (fat) and protein, so drugs that are lipid soluble pass rapidly through the GI membrane. Water-soluble drugs need a carrier, either enzyme or protein, to pass through the membrane. Large particles pass through the cell membrane if they are nonionized (have no positive or negative charge). Weak acid drugs such as aspirin are less ionized in the stomach, and they pass through the stomach lining easily and rapidly. An infant’s gastric secretions have a higher pH (alkaline) than those of adults; therefore, infants can absorb more penicillin. Certain drugs such as calcium carbonate and many of the antifungals need an acidic environment to achieve greater drug absorption; thus food can stimulate the production of gastric acid. Hydrochloric acid destroys some drugs such as penicillin G; therefore a large oral dosage of penicillin is needed to offset the partial dose loss. Drugs administered by many routes do not pass through the GI tract or liver. These include parenteral drugs, eyedrops, eardrops, nasal sprays, respiratory inhalants, transdermal drugs, and sublingual drugs. Remember, drugs that are lipid soluble and nonionized are absorbed faster than water-soluble and ionized drugs.
Blood flow, pain, stress, hunger, fasting, food, and pH affect drug absorption. Poor circulation to the stomach as a result of shock, vasoconstrictor drugs, or disease hampers absorption. Pain, stress, and foods that are solid, hot, or high in fat can slow gastric emptying time, so the drug remains in the stomach longer. Exercise can decrease blood flow by causing more blood to flow to the peripheral muscle, thereby decreasing blood circulation to the GI tract.
Drugs given IM are absorbed faster in muscles that have more blood vessels (e.g., deltoids) than in those that have fewer blood vessels (e.g., gluteals). Subcutaneous tissue has fewer blood vessels, so absorption is slower in such tissue.
Some drugs do not go directly into the systemic circulation following oral absorption but pass from the intestinal lumen to the liver via the portal vein. In the liver, some drugs may be metabolized to an inactive form that may then be excreted, thus reducing the amount of active drug. Some drugs do not undergo metabolism at all in the liver, and others may be metabolized to drug metabolite, which may be equally or more active than the original drug. The process in which the drug passes to the liver first is called the first-pass effect, or hepatic first pass.
Most drugs given orally are affected by first-pass metabolism. Lidocaine and some nitroglycerins are not given orally because they have extensive first-pass metabolism and therefore most of the dose would be destroyed.
Bioavailability is a subcategory of absorption. It is the percentage of the administered drug dose that reaches the systemic circulation. For the oral route of drug administration, bioavailability occurs after absorption and first-pass metabolism. The percentage of bioavailability for the oral route is always less than 100%, but for the IV route it is 100%. Oral drugs that have a high first-pass hepatic metabolism may have a bioavailability of only 20% to 40% on entering systemic circulation. To obtain the desired drug effect, the oral dose could be higher than the drug dose for IV use.
Factors that alter bioavailability include (1) the drug form (e.g., tablet, capsule, sustained-release, liquid, transdermal patch, rectal suppository, inhalation), (2) route of administration (e.g., oral, rectal, topical, parenteral), (3) GI mucosa and motility, (4) food and other drugs, and (5) changes in liver metabolism caused by liver dysfunction or inadequate hepatic blood flow. A decrease in liver function or a decrease in hepatic blood flow can increase the bioavailability of a drug, but only if the drug is metabolized by the liver. Less drug is destroyed by hepatic metabolism in the presence of liver disorder.
With some oral drugs, rapid absorption increases the bioavailability of the drug and can cause an increase in drug concentration. Drug toxicity may result. Slow absorption can limit the bioavailability of the drug, thus causing a decrease in drug serum concentration.
Distribution
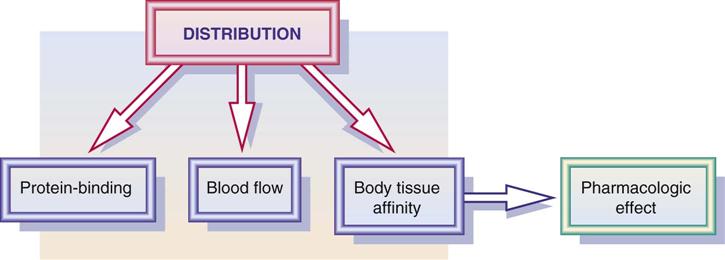
Distribution is the process by which the drug becomes available to body fluids and body tissues. Drug distribution is influenced by blood flow, the drug’s affinity to the tissue, and the protein-binding effect (Figure 1-3). In addition, volume of drug distribution (Vd) is dependent on drug dose and its concentration in the body. Drugs with a larger volume of drug distribution have a longer half-life and stay in the body longer. See the section on metabolism, or biotransformation, later in this chapter.
Protein Binding
As drugs are distributed in the plasma, many are bound to varying degrees (percentages) with protein (primarily albumin). Drugs that are greater than 89% bound to protein are known as highly protein-bound drugs; drugs that are 61% to 89% bound to protein are moderately highly protein-bound; drugs that are 30% to 60% bound to protein are moderately protein-bound; and drugs that are less than 30% bound to protein are low protein-bound drugs. Table 1-1 lists selected highly protein-bound drugs and moderately highly protein-bound drugs. The portion of the drug that is bound is inactive because it is not available to receptors, and the portion that remains unbound is free, active drug. Only free drugs (drugs not bound to protein) are active and can cause a pharmacologic response. As the free drug in the circulation decreases, more bound drug is released from the protein to maintain the balance of free drug. Drugs bound to proteins cannot leave the systemic circulation to get to the site of action. This is why only free drug is active.
TABLE 1-1
PROTEIN-BINDING AND HALF-LIFE OF DRUGS
| DRUG | PROTEIN-BOUND (%) | HALF-LIFE (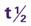 ) (h) ) (h) |
| Highly Protein-Bound Drugs (89%) | ||
| amitriptyline | 97 | 40 |
| chlorpromazine | 95 | 30 |
| diazepam | 98 | 30-80 |
| dicloxacillin | 95 | 0.5-1 |
| furosemide | 95 | 1.5 |
| ibuprofen | 98 | 2-4 |
| lorazepam | 92 | 15 |
| piroxicam | 99 | 30-86 |
| propranolol | 92 | 4 |
| rifampin | 89 | 2 |
| sulfisoxazole | 85-95 | 4.5-7.5 |
| valproic acid | 92 | 15 |
| warfarin | 97 | 20-60 |
| Moderately High Protein-Bound Drugs (61% to 89%) | ||
| erythromycin | 70 | 3 |
| nafcillin | 86 | 2-20 |
| phenytoin | 88 | 10-40 |
| quinidine | 70 | 6 |
| trimethoprim | 70 | 11 |
| Moderately Protein-Bound Drugs (30% to 60%) | ||
| aspirin | 49 | 0.25-2 |
| lidocaine | 50 | 2 |
| meperidine | 56 | 3 |
| pindolol | 40 | 3-4 |
| theophylline | 60 | 9 |
| ticarcillin | 45-65 | 1-1.5 |
| Low Protein-Bound Drugs (30%) | ||
| amikacin | 4-11 | 2-3 |
| amoxicillin | 20 | 1-1.5 |
| atenolol | 6-16 | 6-7 |
| cephalexin | 10-15 | 0.5-1.2 |
| digoxin | 25 | 36 |
| neostigmine bromide | 15-25 | 1-1.5 |
| terbutaline sulfate | 25 | 3-11 |
| timolol maleate | <10 | 3-4 |
| tobramycin sulfate | 10 | 2-3 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


