Chapter 17 Neurology
Basic concepts
1 What is a motor unit? Will most α motor neurons innervate a few or many muscle fibers in a large muscle such as the gluteus maximus?
2 How do upper motor neurons differ from lower motor neurons?
Motor neurons, as the name implies, are neurons involved in stimulating movement. The axons of lower motor neurons (LMNs) (α and γ motor neurons) project directly to skeletal muscle fibers, while upper motor neurons (UMNs) originate in the motor region of the brainstem and cerebral cortex and have no direct contact with skeletal muscles (Fig. 17-1). The cell bodies of UMNs are supraspinal in location, and the axons of these neurons synapse either directly or indirectly, via interneurons, on LMNs located in the ventral horn of the spinal cord or brainstem motor nuclei (e.g., facial nerve motor nucleus).
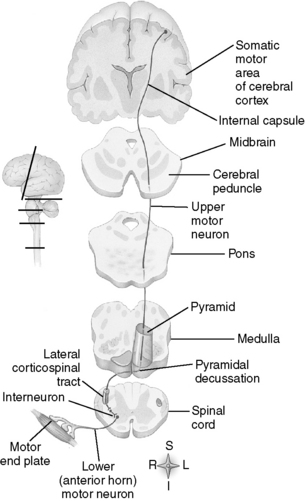
Figure 17-1 Examples of somatic motor pathways.
(From Thibodeau G, Patton K: Anatomy & Physiology, 6th ed. St. Louis, Mosby, 2006.)
UMN lesions produce a different set of symptoms than LMN lesions that you should recognize for boards. While LMN lesions result in flaccid paralysis and weakened reflexes due to decreased contractions from denervated muscles, UMN lesions result in a spastic paralysis and heightened reflexes due to loss of inhibition of γ motor neurons by UMNs. LMN lesions result in fasciculations (random twitches of denervated motor units) and muscle atrophy. UMN lesions are associated with a positive Babinski sign (toes extend upward with plantar stimulation), but LMN lesions are not. UMN and LMN symptoms are explored further in Case 17-1.
5 What are the two ascending sensory pathways and what information does each convey?
1. The anterolateral system, also referred to as the spinothalamic tract, conveys sensations of pain, temperature, and crude (nondiscriminative) touch.
2. The dorsal column–medial lemniscus pathway conveys the sensations of fine touch, vibration, pressure, and conscious proprioception.
Note: Unconscious proprioception is transmitted by the spinocerebellar pathways.
6 What are the two anatomic divisions of the dorsal columns, and from which anatomic structures do these respective divisions relay sensory information?
1. The fasciculus gracilis, which relays information from the lower extremities and from the lower thorax (level T7 and below), is located most medially in the dorsal columns, just as the gracilis muscle is the most medial muscle of the thigh.
2. The fasciculus cuneatus, which relays information from the upper thorax and the upper extremities (levels C2-T6; recall that the C1 spinal nerve provides motor innervation to the muscles of the occipital region) is immediately lateral to the fasciculus gracilis (Fig. 17-2).
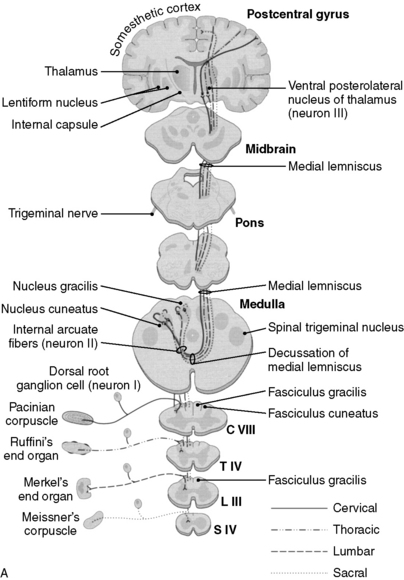
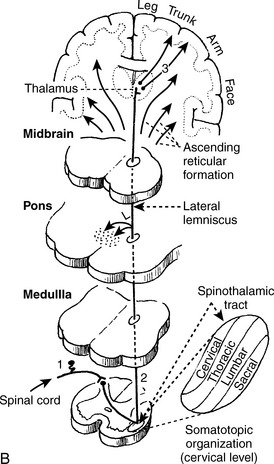
7 At what neuroanatomic locations do projections in the corticospinal tract, dorsal columns, and anterolateral system (spinothalamic system) cross over?
8 Because you know where the major motor and sensory pathways cross over, identify and explain the neurologic deficits that occur in the Brown-Séquard syndrome
The loss of fine touch, vibration, and proprioception (modalities of the dorsal columns) will be on the same side as that of the lesion. This is because the sensory information of the dorsal columns does not cross over until a more superior location (between the brainstem nuclei and the thalamus). The loss of pain and temperature sensation (anterolateral system), however, will be contralateral to the side of the lesion, because the fibers of the anterolateral system ascend and cross over shortly after entering the spinal cord (Fig. 17-3).
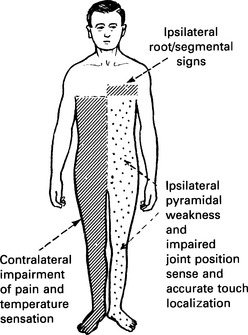
Figure 17-3 Brown-Séquard syndrome.
(From Lindsay KW, Bone I, Callander R [eds]: Neurology and Neurosurgery Illustrated, 3rd ed. Edinburgh, Churchill Livingstone, 2002.)
Note: There may be some loss of all modalities at the level at which the lesion occurs.
9 Where will the motor and sensory deficit manifest (below the head) if there is a lesion of the internal capsule?
3 Why are the signs of hyperreflexia, spastic paralysis, and clonus seen with an upper motor neuron lesion?
4 What lower motor neuron signs are present in this patient?
Note: LMN involvement can be evaluated by electromyography (EMG) and nerve conduction studies.
Case 17-1 continued:
Serum analysis is negative for antibodies to the acetylcholine (ACh) receptor. Cerebrospinal fluid (CSF) analysis is negative for oligoclonal bands (of immunoglobulins), elevated protein, or white blood cells (WBCs). Magnetic resonance imaging (MRI) of the brain and spinal cord appears normal. A muscle biopsy is shown in Figure 17-4.
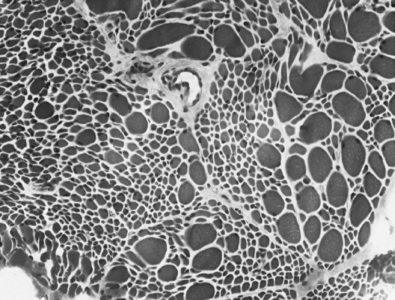
5 What process leads to the findings seen in this biopsy specimen stained with hematoxylin-eosin (H&E)?
9 What are the principal pathologic findings in amyotrophic lateral sclerosis?
There is loss of pyramidal cells in the motor cortex, leading to fibrosis (which generally manifests in the central nervous system [CNS] as astrocytic gliosis) of the lateral corticospinal tracts. In addition, there is loss of ventral horn neurons throughout the length of the spinal cord, resulting in thinning of ventral (motor) nerve roots. The affected muscles show denervation atrophy with (muscle) fiber type grouping upon reinnervation. There usually is sparing of sensory tracts and cognitive function, which explains why this patient’s mental status and sensory function are both completely normal. Extraocular muscles also are often spared, leaving some patients with severe disease progression no means of communication other than eye movements (Fig. 17-5).
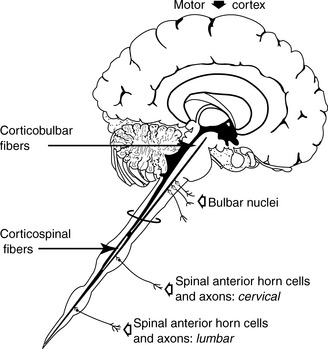
Figure 17-5 Sites of the lesions in amyotrophic lateral sclerosis (ALS).
(From Pryse-Phillips WM, Murray TJ: Essential Neurology: A Concise Textbook. New York, Medical Examination Publishing, 1992.)
12 Why is amyotrophic lateral sclerosis often confused for syringomyelia and vice versa?
Summary Box: Amyotrophic Lateral Sclerosis
 Clinical presentation is one of chronic degeneration of upper and lower motor neurons, generally without sensory or cognitive involvement.
Clinical presentation is one of chronic degeneration of upper and lower motor neurons, generally without sensory or cognitive involvement.

 Muscle biopsy shows type grouping and group atrophy of muscle fibers.
Muscle biopsy shows type grouping and group atrophy of muscle fibers.
 Most cases are acquired; one familial form is associated with superoxide dismutase 1 mutations.
Most cases are acquired; one familial form is associated with superoxide dismutase 1 mutations.

5 Why should we determine whether this patient is taking medications such as haloperidol or metoclopramide?
6 What cerebral structures are affected in Parkinson’s disease, and how does this play into the bradykinesia and akinesia observed?
In Parkinson’s disease, the dopaminergic, neuromelanin-containing neurons in the substantia nigra selectively degenerate over time. These neurons normally project to the basal ganglia via the nigrostriatal tract. The basal ganglia then influence execution of learned motor plans by modulating signals between the thalamus and motor cortex. Within the basal ganglia, there are two pathways leading to output to the thalamus: the direct and indirect pathways. Activation of the direct pathway facilitates desired movement by stimulating the thalamus via inhibition of the globus pallidus internus and substantia nigra pars reticularis (both of which normally inhibit the thalamus), whereas activation of the indirect pathway inhibits unwanted movement by inhibiting the thalamus. Nigrostriatal dopaminergic inputs activate the direct pathway and inhibit the indirect pathway, thus stimulating motion. Decreased dopamine levels in Parkinson’s disease manifest with a net decrease in motor activity. However, this model does not yet account for the patient’s tremor. Because dopamine decreases release of ACh, a decrease in dopamine levels leads to a relative excess of striatal ACh in patients with Parkinson’s disease. ACh opposes the actions of dopamine and activates the indirect pathway, which disinhibits suppression of unwanted movements and results in the pill-rolling tremor that is characteristic of the disease. It is important that you understand the imbalance of dopamine and ACh levels in Parkinson’s patients because dopamine agonists and anticholinergics are useful in treating this disease (Fig. 17-6).
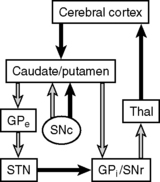
7 What medication did you start the patient on, and why is it, rather than dopamine, used to treat Parkinson’s disease?
9 Drugs such as bromocriptine and pergolide are also used to treat Parkinson’s disease. How do they exert their effects?
11 Why is it preferable to selectively inhibit monoamine oxidase B, rather than both monoamine oxidase A and monoamine oxidase B, in Parkinson’s disease?
14 How does the drug MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) cause parkinsonism, and is this a reversible process?
15 Why should you be suspicious of a diagnosis of Parkinson’s disease in a patient being treated for schizophrenia?
16 What would a pathologist look for to establish the diagnosis of Parkinson’s disease in evaluation of the brain at autopsy?
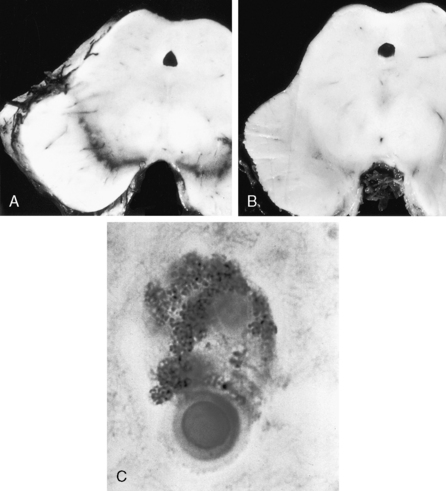
 Bilateral depigmentation of the midbrain substantia nigra, due to loss of dopaminergic, neuromelanin-containing neurons (Fig. 17-7A [normal] and B [Parkinson’s])
Bilateral depigmentation of the midbrain substantia nigra, due to loss of dopaminergic, neuromelanin-containing neurons (Fig. 17-7A [normal] and B [Parkinson’s])
 The presence of neuronal Lewy bodies in degenerated substantia nigra neurons (eosinophilic, cytoplasmic inclusions containing α-synuclein and ubiquitin) (Fig. 17-7C)
The presence of neuronal Lewy bodies in degenerated substantia nigra neurons (eosinophilic, cytoplasmic inclusions containing α-synuclein and ubiquitin) (Fig. 17-7C)
Summary Box: Parkinson’s Disease




4 If a mini-mental status examination shows the patient to be mildly demented, with deficits in organization, concentration, and short-term memory, what is the most likely diagnosis?
5 What pathologic lesion would be visible on imaging?
MRI of the head is notable for significant atrophy of the basal ganglia, especially the caudate nucleus. In fact, Huntington’s disease is caused by degeneration of γ-aminobutyric acid (GABA) neurons belonging to the indirect pathway in the caudate nucleus. This is significant because the striatal nuclei are the main inhibitors of undesirable movement. The ventricular enlargement that is often apparent on autopsy (Fig. 17-8, right) is due to loss of neurons in the basal ganglia.
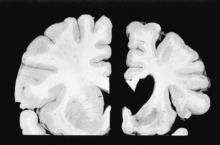
6 What is the mode of inheritance?
Huntington’s disease is inherited in an autosomal dominant manner.
7 What neurotransmitter is reduced in the basal ganglia in Huntington’s disease and how does this relate to the hyperkinetic motor abnormalities seen in Huntington’s disease?
8 What does the term “penetrance” imply with respect to genetic diseases, and is the penetrance of Huntington’s disease high or low?
9 What is the meaning of anticipation with respect to genetic diseases and what is the cause of anticipation in Huntington’s disease?
11 How is it possible for someone with a negative family history of Huntington’s disease to develop the disease?
12 Why might it make sense to measure levels of serum ceruloplasmin in patients who present with similar motor abnormalities and a similar family history?
Summary Box: Huntington’s Disease




1 Given her diplopia, which cranial nerves should you examine particularly carefully?
Note: Diplopia and other ocular pathologic conditions are discussed in further detail in Chapter 18.
2 Why would an edrophonium chloride (Tensilon) test help determine whether the symptoms are of nerve, neuromuscular junction, or muscle origin?
3 If computed tomography and magnetic resonance imaging scans of the chest were ordered, what diagnosis would be supported by finding a thymoma?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


