Chapter 3 Nephrology
Clinical renal disease
2 What are the three etiologic classifications of acute renal failure?
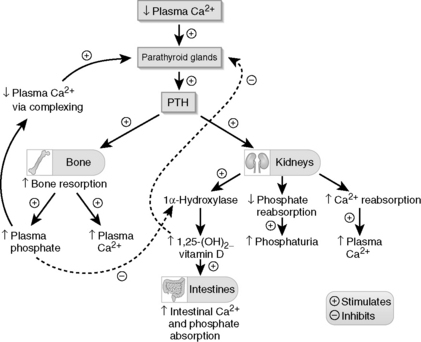
Renal failure is classified as prerenal, renal (intrinsic), or postrenal (obstructive). This man’s renal failure clearly has an obstructive etiology. Acute bladder distention caused by long-standing BPH has resulted in bilateral hydronephrosis (dilated renal pelves) and acute renal failure. Figure 3-1 shows severe hydronephrosis from long-standing obstruction, with marked dilatation of the renal pelvis and calyces and loss of cortical parenchyma.
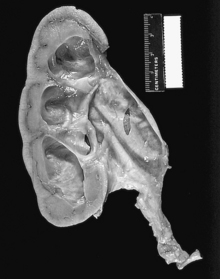
3 How is glomerular filtration rate (GFR) calculated?
The GFR can be calculated using the following formula:
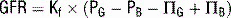
where
5 Why was it important to ask this patient about tricyclic antidepressants, antipsychotics, antihistamines, and sympathomimetics?
Summary Box: Obstructive Acute Renal Failure



3 What is the cause of the decreased glomerular filtration rate and oliguria seen in acute tubular necrosis?
5 Other than the presence of “muddy brown” casts, how can prerenal azotemia due to ischemia be differentiated from ischemic acute tubular necrosis?
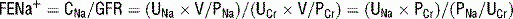
where
CX = clearance of substance X; volume of plasma from which substance X is cleared per unit time
UX = excretion rate of substance X
PX = plasma concentration of substance X
Related Question
6 What is rhabdomyolysis and how can it cause acute tubular necrosis?



2 What are the three major types of NSAID-induced renal toxicity?
NSAIDs can cause a bewildering array of renal side effects.
Summary Box: Acute Interstitial Nephritis



1 What is most likely causing her blood urea nitrogen and creatinine elevation?
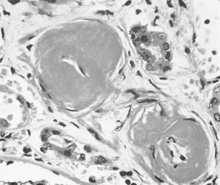
The pathologic changes associated with longstanding hypertension are termed hypertensive nephrosclerosis (also known as benign nephrosclerosis or hyaline arteriolar nephrosclerosis). As shown in Figure 3-2, hyaline arteriosclerosis is associated with hyaline deposition, marked thickening of the walls, and a narrowed lumen.
2 What is the value of the normal fasting glucose and hemoglobin A1c levels in the differential diagnosis?
3 What is the difficulty in establishing that this patient’s renal failure was definitely due to hypertension, even if no other specific disease processes can be identified on renal biopsy?
6 How does parathyroid hormone normally act to regulate serum Ca2+? How are parathyroid hormone levels normally regulated?
In general, PTH release is stimulated by low Ca2+ levels, whereas its release is inhibited (in a negative feedback mechanism) by the active 1,25-dihydroxy form of vitamin D (Fig. 3-3).