Chapter 11 Genetic and Metabolic Disease
Basic Concepts
1 What is an enzymopathy and how does it result in clinical symptoms?
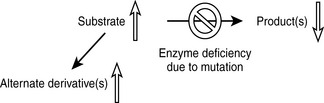
An enzymopathy is a genetic disease in which a deficiency in activity of an enzyme leads to a block in a metabolic pathway. The altered (usually reduced) enzymatic activity can be due to reduced cellular expression of the enzyme or to expression of a dysfunctional enzyme. The pathologic manifestations of the enzyme deficiency are a result of the accumulation of substrate (or its derivatives) prior to the blockage, a lack of the product(s), or a combination of both (Fig. 11-1).
3 Explain why the pathologic consequences of X-linked enzymopathies are manifested almost exclusively in males
The USMLE loves to ask students to calculate inheritance risk. This will require you to know the inheritance pattern of the disease in question and apply it to the Hardy-Weinberg principle. Recall that if a population is in Hardy-Weinberg equilibrium, and p is the frequency of the normal allele while q is the frequency of the abnormal allele, disease prevalence = p2 + 2pq + q2 where p2 and q2 represent the prevalence of homozygosity and 2pq is heterozygosity prevalence (see Case 11-2, q4).
4 What is the process of lyonization and how may it cause the manifestation of X-linked diseases in females?
5 Why do some diseases show an autosomal dominant pattern of inheritance? Why do the genetic diseases of connective tissue usually fall within this category?
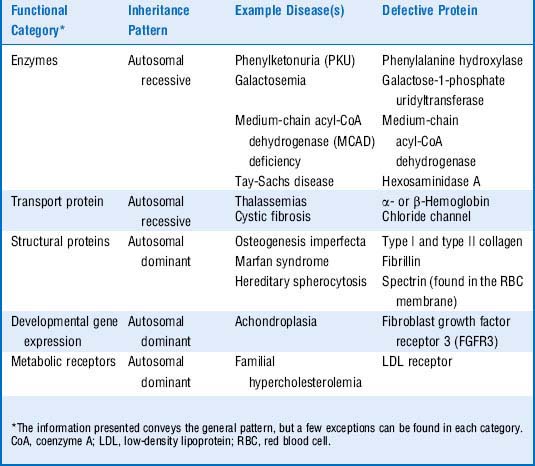
6 What is the general relationship between the function of a protein and its pattern of inheritance?
Table 11-1 shows these relationships.
7 What are the following molecular biology diagnostic methods used for? Explain briefly how they work
Summary Box: Basic Concepts in Biogenetics
 Enzymopathies (enzyme deficiencies) are caused by a deficiency in activity of an enzyme, resulting in a toxic accumulation of intermediates, a lack of products, or both. Enzymopathies are usually autosomal recessive or X-linked recessive.
Enzymopathies (enzyme deficiencies) are caused by a deficiency in activity of an enzyme, resulting in a toxic accumulation of intermediates, a lack of products, or both. Enzymopathies are usually autosomal recessive or X-linked recessive.

 Defects in structural proteins typically exhibit autosomal dominant inheritance.
Defects in structural proteins typically exhibit autosomal dominant inheritance.

2 What is the major defect and underlying pathophysiology of this disorder?
The pathology is primarily a result of substrate (phenylalanine) accumulation, which causes severe neuronal damage, mental retardation, growth retardation, and motor dysfunction. The lack of neurotransmitter compounds derived from tyrosine (particularly the catecholamines dopamine, norepinephrine, and epinephrine) may also contribute to damage of the central nervous system (CNS). Other manifestations include a predisposition to eczema, a “musty” odor (caused by phenylketone excretion into sweat), and fair skin coloring (due to tyrosine deficiency, which normally serves as a precursor to melanin) (Fig. 11-2).
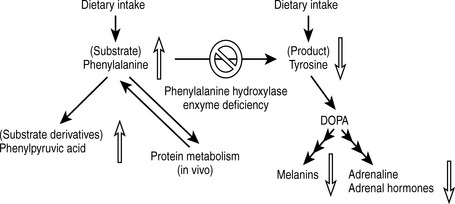
Figure 11-2 Pathologic mechanisms of phenylketonuria.
(From Brown TA, Brown D: USMLE Step 1 Secrets. Philadelphia, Hanley & Belfus, 2004.)
4 Given the fact that phenylketonuria is a relatively rare condition (prevalence rates range from 1 in 2600 to 1 in 200,000 live births), why does it make sense to screen all neonates for this condition?
Related question
6 Why is screening for congenital hypothyroidism (cretinism), congenital adrenal hyperplasia, and galactosemia also routinely performed in newborns?
Summary Box: Phenylketonuria and Neonatal Screening




1 If the parents decide to have another child, what is the probability of that child having cystic fibrosis?
2 If the parents want to have another child, what kind of genetic screening methods are available for them to consider?
A few genetic screening methods are available:
3 Despite having mutations in the same gene, why do patients with cystic fibrosis exhibit significant variability in disease severity?
4 Assuming a cystic fibrosis prevalence rate of 1 in 2500, what is the carrier frequency for this disease?
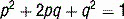
p = frequency of normal allele
q = frequency of abnormal allele (1 − p)
p2 = frequency of unaffected individuals
2pq = frequency of carriers (usually asymptomatic in autosomal recessive diseases)
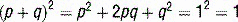
Summary Box: Population Genetics and Prenatal Genetic Screening



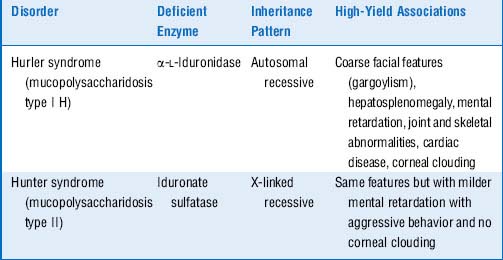
5 What are the mucopolysaccharidoses?
Mucopolysaccharidoses are a different type of lysosomal storage disease in which the substrates that accumulate in the lysosomes are extracellular matrix molecules called glycosaminoglycans (which were previously known as mucopolysaccharides). Like the sphingolipidoses, these diseases are caused by hereditary deficiency of lysosomal enzymes. The two main examples of this type of disease, Hurler syndrome and the similar but less severe Hunter syndrome, are both caused by accumulation of the glycosaminoglycans heparan sulfate and dermatan sulfate (Table 11-2).
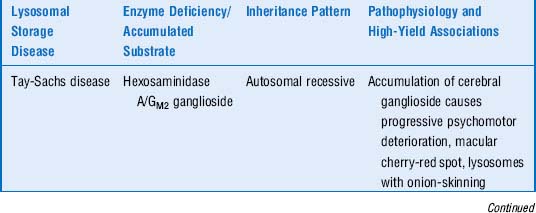
6 Quick review: Cover the three columns on the right side of Table 11-3 and attempt to describe the enzyme deficiency, accumulated substrate, inheritance pattern, pathophysiology, and any high-yield associations for the listed lysosomal storage disorders


Summary Box: Lysosomal Storage Diseases




2 What is the normal function of the purine “salvage” pathway?
The purine salvage pathway functions to “salvage” purine metabolites such as hypoxanthine and guanine, preventing them from being unnecessarily degraded and then renally excreted as uric acid. (Hypoxanthine is another purine that is an intermediate in the synthesis or degradation of adenosine monophosphate [AMP] or guanosine monophosphate [GMP]; Fig. 11-3). As shown in Figure 11-3, the salvage pathway recycles these metabolites to replenish the purine bases guanine and adenine by the action of the HGPRT enzyme. Normally, the de novo pathway (smaller dark arrows) provides only about 10% of the daily purine requirement, whereas the salvage pathway (large curved arrows) provides the remaining 90%. The amount of net degradation to uric acid (open arrows) is always balanced with the amount of purines synthesized via the de novo pathway. It follows that the loss of the salvage pathway would result in a dramatic increase in de novo purine synthesis and a similarly dramatic increase in uric acid generation.
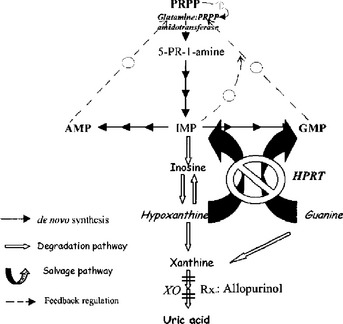
3 How do defects in the purine salvage pathway cause hyperuricemia?
In LNS, HGPRT activity is less than 1% of normal. Owing to the absence of HGPRT, the ability to reutilize hypoxanthine and guanine to make the purine nucleotides inosine monophosphate (IMP) and GMP is lost, so these intermediates are degraded to uric acid. As explained already in the preceding question, the purine requirements in this case must be met by increased de novo synthesis. Additionally, because of the reduced levels of IMP and GMP, the feedback inhibition normally exerted by IMP and GMP upon the de novo pathway is lost (broken arrows in Fig. 11-3), even further promoting the activity of the de novo synthesis pathway. Because purine synthesis via the de novo pathway must be balanced by purine degradation into to uric acid, the dramatic increase in de novo synthesis results in severe hyperuricemia.
6 How might this patient be managed pharmacologically?
Allopurinol is useful in the treatment of hyperuricemia of any cause. It works by preventing uric acid production by inhibiting the enzyme xanthine oxidase (XO) (see Fig. 11-3). The xanthine and hypoxanthine that accumulate instead are more soluble and readily excreted than uric acid.
Summary Box: Lesch-Nyhan Syndrome and Purine Metabolism



3 How is glycogen normally synthesized and degraded in the liver?
Glycogen degradation is primarily dependent on the activity of the enzyme glycogen phosphorylase (Fig. 11-4).
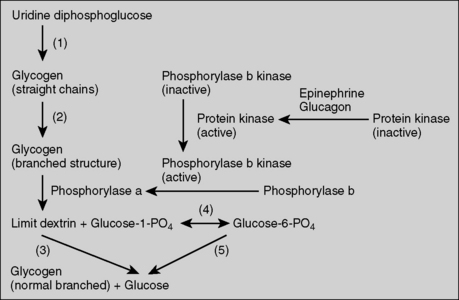
6 What is the explanation for his severe fasting hypoglycemia and lactic acidosis?
During short-term fasting, liver glycogenolysis is the major pathway that maintains blood glucose. When G6Pase is deficient, glycogenolysis is not effective at releasing glucose into the bloodstream, leading to hypoglycemia. In addition, the release of glucose made by gluconeogenesis during fasting is also impaired because this process is also dependent upon the enzyme G6Pase. This further contributes to hypoglycemia. Excessive accumulation of G6P greatly promotes glycolysis, resulting in high levels of pyruvate production. The pyruvate is then converted into lactate when the mitochondrial uptake of pyruvate is saturated, resulting in lactic acidosis (see Fig. 11-4).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


