Biologic Response Modifiers
Objectives
Key Terms
absolute neutrophil count, p. 561
biologic response modifiers, p. 555
colony-stimulating factors, p. 557
dose-limiting toxicity, p. 556
erythropoietin, p. 558
erythropoietin-stimulating agents, p. 558
granulocyte, p. 560
granulocyte colony-stimulating factor, p. 560
granulocyte-macrophage colony-stimulating factor, p. 562
hybridoma technology, p. 555
interferons, p. 556
interleukins, p. 565
keratinocyte growth factor, p. 566
lymphokines, p. 565
macrophages, p. 556
myelosuppression, p. 557
nadir, p. 561
neutrophils, p. 557
pegylation, p. 562
recombinant DNA, p. 555
thrombocytopenia, p. 557
The authors gratefully acknowledge the work of Carolee A. Polek, who updated this chapter for the eighth edition.
 http://evolve.elsevier.com/KeeHayes/pharmacology/
http://evolve.elsevier.com/KeeHayes/pharmacology/
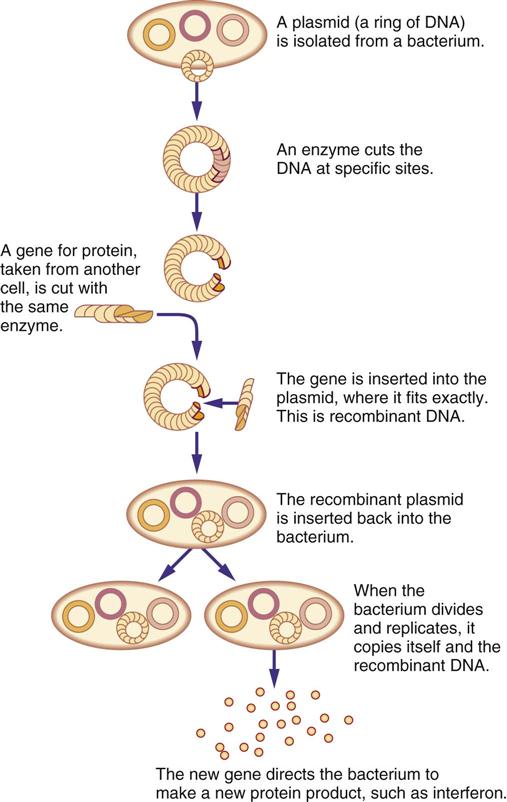
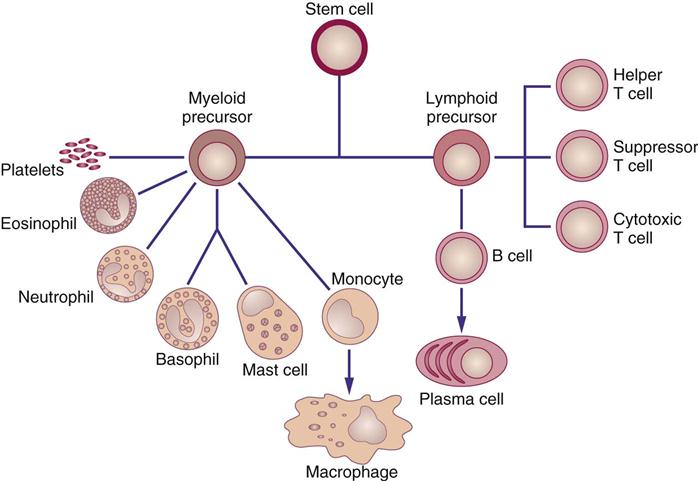
Biologic response modifiers (BRMs) are a class of pharmacologic agents used to enhance the body’s immune system. (Another group of immunostimulants are monoclonal antibodies, considered targeted therapy agents, which are discussed in Chapter 38.) Recombinant DNA (the genetic engineering process that produces mass quantities of human proteins) and hybridoma technology (the process that uses mice to mass-produce monoclonal antibodies) are two advances that have led to commercial mass-production of BRMs (Figure 39-1). Interferons (α, β, γ), colony-stimulating factors, interleukins, and monoclonal antibodies are some currently known BRMs. With the exception of monoclonal antibodies, BRMs are complex proteins produced by the cells of the immune system (Figure 39-2). Individuals taking BRMs are immunocompromised and should be counseled on concurrent drug and herb use (Herbal Alert 39-1).
 Herbal Alert 39-1
Herbal Alert 39-1
Biologic Response Modifiers
Herbal preparations are generally not recommended for patients receiving BRMs. Several herbs (e.g., elderberry, echinacea, ginseng, goldenseal, green tea) act to stimulate the immune system. However, patients who take BRMs are immunocompromised, and taking any herbal preparations in combination is not recommended.
Three functions of BRM have been identified:
• Enhance host immunologic function (immunomodulation)
• Destroy or interfere with tumor activities (cytotoxic/cytostatic effects)
• Promote differentiation of stem cells (other biologic effects) (see Figure 39-2).
New BMR drugs are always under development and are being investigated for clinical effectiveness. Several categories of these drugs have been approved by the U.S. Food and Drug Administration (FDA) and are commercially available: interferons, colony-stimulating factors (erythropoietin-stimulating agents, granulocyte colony-stimulating factors, granulocyte macrophage colony-stimulating factors, thrombopoietic growth factors), and interleukin-2. In addition, a number of monoclonal antibodies have been approved by the FDA (see Chapter 38). Interferons, colony-stimulating factors, and interleukin-2 are discussed in this chapter.
Interferons
Interferons (IFNs) are a family of naturally occurring proteins that were first discovered in the 1950s. Three major types of IFNs have been identified: alpha (α) IFN, beta (β) IFN, and gamma (γ) IFN. Each type is produced by a different cell within the immune system. All three types can be manufactured using recombinant DNA technology. Type I IFNs include IFN-α and IFN-β, which bind selectively to either α- or β-cell surface receptors on effector cells. Interferon gamma (IFN-γ) is a type II IFN and binds to a different cell surface receptor. Common to both Type I and II IFNs are their abilities to (1) regulate the immune system to improve resistance to invading microorganisms and (2) reduce cell proliferation. However, each IFN group has specific functionality as well.
Interferon Alpha
B-cells, T-cells, macrophages (type of monocyte—the major phagocytic cells of the immune system), and null cells produce interferon alpha (IFN-α) (Roferon-A, Intron A) in response to the presence of viruses or tumor cells. This interferon has been shown to have antiviral, antiproliferative, and immunomodulatory effects, which means that IFN-α inhibits intracellular replication of viral DNA, interferes with tumor cell growth, and enhances natural killer (NK) cell (antitumor) activity. Recombinant IFN-α is manufactured as Roferon-A (alpha-2a) and Intron A (alpha-2b).
Roferon-A is used in the treatment of (1) hairy cell leukemia; (2) chronic myelogenous leukemia (CML), and (3) AIDS-related Kaposi’s sarcoma. Intron A is used in the treatment of (1) malignant melanoma, (2) hairy cell leukemia, (3) follicular non-Hodgkin’s lymphoma (NHL), (4) AIDS-related Kaposi sarcoma, and (5) nonmalignant chronic hepatitis B and C and condylomata acuminata. Clinically IFN-α has been used to treat non-Hodgkin’s lymphoma (intermediate grade), cutaneous T-cell lymphoma, multiple myeloma, renal cell carcinoma, bladder cancer, and carcinoid (a type of cancer-like disease). Intravesical administration of IFN-α has proved successful for low-grade bladder tumors; intraperitoneal administration of IFN-α has been used to treat ovarian cancer; and intralesional application of IFN-α has been used to treat melanoma and basal cell carcinoma. IFN-α-n3 is a human leukocyte-derived interferon that binds to the same receptors as IFN-α-2b. It is FDA approved for intralesional treatment of refractory or recurring external condylomata acuminata.
Interferon Beta
Interferon beta (IFN-β) (Betaseron, IFN-β-1b) is indicated for the treatment of multiple sclerosis. Specifically this interferon (1) enhances the activity of suppressor T-cells (thymocyte-dependent lymphocytes that turn off an immune reaction—an active part of the cell-mediated immune system), (2) reduces the production of proinflammatory cytokines, (3) reduces antigen-presentation (recruiting members of the immune system to start an immune reaction), and (4) inhibits the movement of lymphocytes in the central nervous system.
Interferon Gamma
Interferon gamma (IFN-γ) (Actimmune, IFN-γ-1b) is indicated for (1) delaying time to disease progression in patients with severe malignant osteopetrosis, (2) reducing the frequency and severity of serious infections related to chronic granulomatous disease, and (3) treating patients with chronic myelogenous leukemia and renal cell carcinoma. IFN-γ-1b has been shown to (1) enhance oxidative metabolism of macrophages, (2) enhance antibody-dependent cell cytotoxicity (ADCC), (3) activate natural killer (NK) cells, and (4) enhance Fc receptors (on antibody) and major histocompatibility antigens (HLA). Malignant osteopetrosis is a congenital disorder hallmarked by two characteristic defects: a defect in osteoclasts that causes bone overgrowth and a defect in phagocyte oxidative metabolism, which can be improved with IFN-γ-1b. Chronic granulomatous disease (CGD) is an inherited error in leukocyte function related to enzyme defects that are responsible for killing invading microorganisms (phagocyte superoxide generation).
Pharmacokinetics
IFN-α is metabolized by the liver and filtered by the kidneys. However, the body absorbs approximately 80% of the dose. Peak serum concentrations are reached 4 to 8 hours after administration. IFN-α can be administered subcutaneously (subQ), intramuscularly (IM), and intravenously (IV), although subQ and IM are the preferred routes of administration; subQ administration is recommended for patients with platelet counts below 50,000. Interferons-α-n3, -β and -γ are not discussed further because they are not used as antineoplastic agents.
Side Effects and Adverse Reactions
The major side effect of IFN-α is a flulike syndrome. Other effects occur in the gastrointestinal (GI), neurologic, cardiopulmonary, renal, hepatic, hematologic, and dermatologic systems.
The flulike syndrome is characterized by chills, fever, fatigue, malaise, and myalgias (muscle aches). Chills can occur 3 to 6 hours after IFN-α administration and may progress to rigors. A fever as high as 101° F to 104° F (39° C to 40° C) may occur within 30 to 90 minutes after the onset of chills and last 24 hours. Fatigue, malaise, and myalgias are cumulative side effects; fatigue is the dose-limiting toxicity (i.e., the side effect that necessitates a decrease in dose or discontinuation of the drug).
GI side effects include nausea, diarrhea, vomiting, anorexia, taste alterations, and xerostomia (dry mouth). These side effects are mild; anorexia is considered the dose-limiting toxicity for this system.
Neurologic side effects are reversible (after the drug is stopped) and occur in 70% of patients who receive IFN-α. These effects are manifested by mild confusion, somnolence (sleepiness), irritability, poor concentration, seizures, transient aphasia (temporary loss of ability to speak), hallucinations, paranoia, and psychoses. Depression, suicidal ideation, and suicide attempts have occurred in patients taking IFN-α, requiring drug discontinuance.
Cardiopulmonary side effects are dose-related and occur more frequently in older adults and patients with underlying cardiac disease. Side effects include tachycardia, pallor, cyanosis, tachypnea, nonspecific electrocardiographic changes, rare myocardial infarction, and orthostatic hypotension.
Renal and hepatic effects are dose-dependent and usually result in few or no symptoms. The effects are manifested by increased blood urea nitrogen (BUN) and creatinine levels, proteinuria, and elevated transaminases.
Hematologic effects are reversible and dose-limiting. Neutropenia (decreased number of neutrophils in the blood) and thrombocytopenia (decreased number of thrombocytes in the blood) are manifestations of such effects. Neutropenia is usually rare and does not predispose the patient to infection. Thrombocytopenia is more common in patients with hematopoietic (affecting the formation of blood cells) diseases than in those with solid tumors.
Maculopapular rashes of the trunk and extremities, pruritus, irritation at the injection site, desquamation (shedding of epithelial cells of the skin), and alopecia are dermatologic effects of IFN-α. Alopecia can occur after more than 4 months of therapy.
Dosing and Preparation
Roferon-A is supplied in prefilled syringes containing 3 million International Units (IU) per 0.5 mL, 6 million IU per 0.5 mL, and 9 million IU per 0.5 mL. All prefilled syringes must be refrigerated at 36° F to 46° F (2° C to 8° C). The syringes should not be frozen or shaken and must be protected from light.
Intron A is supplied as single-use vials (either powder or liquid), multidose vials, and multidose pens. The lyophilized powder is for single-dose use and is available in 10, 18, and 50 million IU. The powder is reconstituted with 1 mL of the provided diluent (sterile water for injection) and gently swirled to dissolve. Final vial concentrations are 10 million IU/mL, 18 million IU/mL, and 50 million IU/mL. The single-dose solution vial for injection is available as 10 million IU (10 million IU/mL). Multidose vials are available as 18 million IU (3 million IU/0.5 mL) and 25 million IU (5 million IU/0.5 mL). Multidose pens for injection are available containing 3 million IU (concentration 22.5 million IU/1.5 mL, delivering 3 million IU/0.2 mL, and containing 6 doses), 5 million IU (concentration 37.5 million IU/1.5 mL, delivering 5 million IU/0.2 mL, and containing 6 doses), and 10 million IU (concentration 75 million IU/1.5 mL, delivering 10 million IU/0.2 mL, and containing 6 doses). Multidose solutions and pens must be stored at 36° F to 46° F (2° C to 8° C). The IFN alphas are listed in Table 39-1 with their dosages, uses, and considerations.
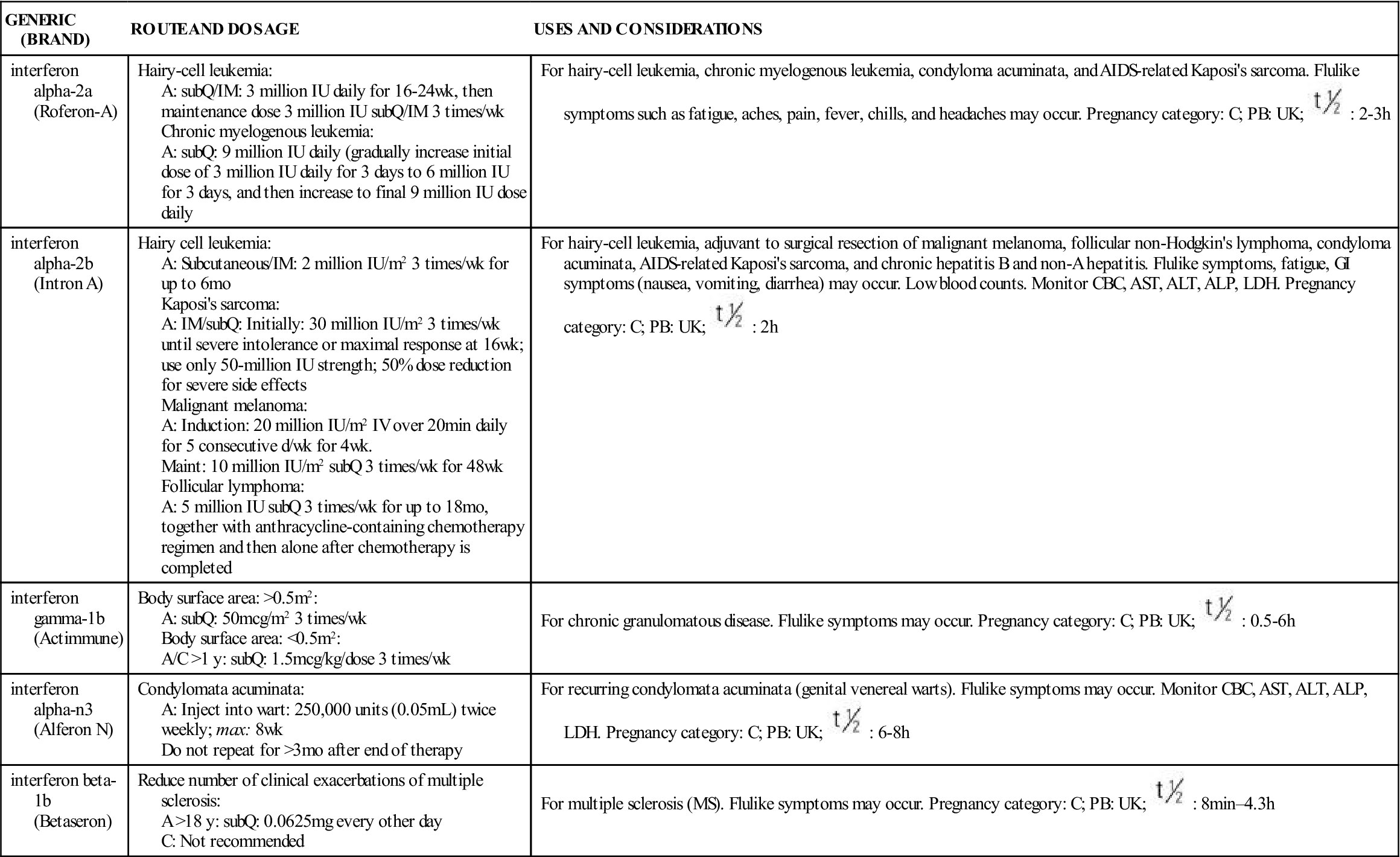
TABLE 39-1
BIOLOGIC RESPONSE MODIFIERS: INTERFERONS
| GENERIC (BRAND) | ROUTE AND DOSAGE | USES AND CONSIDERATIONS |
| interferon alpha-2a (Roferon-A) | Hairy-cell leukemia: A: subQ/IM: 3 million IU daily for 16-24 wk, then maintenance dose 3 million IU subQ/IM 3 times/wk Chronic myelogenous leukemia: A: subQ: 9 million IU daily (gradually increase initial dose of 3 million IU daily for 3 days to 6 million IU for 3 days, and then increase to final 9 million IU dose daily | For hairy-cell leukemia, chronic myelogenous leukemia, condyloma acuminata, and AIDS-related Kaposi’s sarcoma. Flulike symptoms such as fatigue, aches, pain, fever, chills, and headaches may occur. Pregnancy category: C; PB: UK;  : 2-3 h : 2-3 h |
| interferon alpha-2b (Intron A) | Hairy cell leukemia: A: Subcutaneous/IM: 2 million IU/m2 3 times/wk for up to 6 mo Kaposi’s sarcoma: A: IM/subQ: Initially: 30 million IU/m2 3 times/wk until severe intolerance or maximal response at 16 wk; use only 50-million IU strength; 50% dose reduction for severe side effects Malignant melanoma: A: Induction: 20 million IU/m2 IV over 20 min daily for 5 consecutive d/wk for 4 wk. Maint: 10 million IU/m2 subQ 3 times/wk for 48 wk Follicular lymphoma: A: 5 million IU subQ 3 times/wk for up to 18 mo, together with anthracycline-containing chemotherapy regimen and then alone after chemotherapy is completed | For hairy-cell leukemia, adjuvant to surgical resection of malignant melanoma, follicular non-Hodgkin’s lymphoma, condyloma acuminata, AIDS-related Kaposi’s sarcoma, and chronic hepatitis B and non-A hepatitis. Flulike symptoms, fatigue, GI symptoms (nausea, vomiting, diarrhea) may occur. Low blood counts. Monitor CBC, AST, ALT, ALP, LDH. Pregnancy category: C; PB: UK;  : 2 h : 2 h |
| interferon gamma-1b (Actimmune) | Body surface area: >0.5 m2: A: subQ: 50 mcg/m2 3 times/wk Body surface area: <0.5 m2: A/C >1 y: subQ: 1.5 mcg/kg/dose 3 times/wk | For chronic granulomatous disease. Flulike symptoms may occur. Pregnancy category: C; PB: UK; 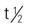 : 0.5-6 h : 0.5-6 h |
| interferon alpha-n3 (Alferon N) | Condylomata acuminata: A: Inject into wart: 250,000 units (0.05 mL) twice weekly; max: 8 wk Do not repeat for >3 mo after end of therapy | For recurring condylomata acuminata (genital venereal warts). Flulike symptoms may occur. Monitor CBC, AST, ALT, ALP, LDH. Pregnancy category: C; PB: UK; 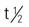 : 6-8 h : 6-8 h |
| interferon beta-1b (Betaseron) | Reduce number of clinical exacerbations of multiple sclerosis: A >18 y: subQ: 0.0625 mg every other day C: Not recommended | For multiple sclerosis (MS). Flulike symptoms may occur. Pregnancy category: C; PB: UK; 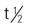 : 8 min–4.3 h : 8 min–4.3 h |
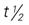 , half-life; UK, unknown; wk, week; y, year; >, greater than; <, less than.
, half-life; UK, unknown; wk, week; y, year; >, greater than; <, less than.Administration
The administration of IFN-α is contraindicated in patients with a known hypersensitivity to IFN-α, mouse immunoglobulin, or any component of the product. It should be used cautiously in patients with severe cardiac, renal, or hepatic disease and in those with seizure disorders or central nervous system dysfunction. Manufacturers recommend administering the agent to persons 18 years or older only under the supervision of a qualified physician. Women of childbearing age should use contraceptives while receiving IFN-α because of the agent’s effects on serum estradiol and progesterone concentrations. Studies have not demonstrated fertility or teratogenic effects in men.
It is recommended that baseline and periodic complete blood counts (CBCs) and liver function tests be performed during the course of IFN therapy. Manufacturers suggest that patients undergo IFN therapy for at least 6 months before a decision is made to continue treatment in those who respond or discontinue treatment in those who do not; optimal treatment duration has not been determined. Patients should be well hydrated during IFN therapy, especially during treatment for malignant melanoma, because this minimizes severe side effects. Dose reductions of 50% or drug discontinuation should be considered if severe adverse reactions occur. Concurrent or prior treatment with chemotherapeutic agents or radiation therapy may increase the effectiveness and toxicity of IFN-α.
Colony-Stimulating Factors
Hematopoietic colony-stimulating factors (CSFs) are proteins that stimulate or regulate the growth, maturation, and differentiation of bone marrow stem cells. CSFs are manufactured through recombinant DNA techniques.
Although CSFs are not directly tumoricidal, they are useful in cancer treatment, because they do the following:
• Reduce bone marrow recovery time after bone marrow transplantation
• Enhance macrophage or granulocyte tumor-, virus-, and fungus-destroying ability
• Prevent severe thrombocytopenia after myelosuppressive chemotherapy
CSFs have been used to treat patients with neutropenia secondary to disease or treatment and can be administered both IV and subQ. The CSFs that are FDA approved for clinical use are erythropoietin-stimulating agents, granulocyte colony-stimulating factor, granulocyte macrophage colony-stimulating factor, and thrombopoietic growth factor.
Erythropoietin-Stimulating Agents
Erythropoietin-stimulating agents (ESAs) include epoetin alpha (Procrit, Epogen) and darbepoetin alpha (Aranesp). Erythropoietin is a glycoprotein produced by the kidney; it stimulates red blood cell (RBC) production in response to hypoxia (decreased oxygen to body tissues). Specifically, erythropoietin stimulates the division and differentiation of committed RBC progenitors (parent cells destined to become circulating RBCs) in the bone marrow. ESAs have undergone close scrutiny as data from clinical trials reveal more deaths in cancer patients receiving ESAs when compared to patients receiving RBC transfusions. Therefore ESAs must be used cautiously and only as indicated, because of the increased risk for death and serious cardiovascular events when administered to increase hemoglobin levels to >12 g/dL. Procrit is currently FDA approved for treatment of anemia secondary to chronic renal failure (CRF), zidovudine (AZT)-treated human immunodeficiency virus (HIV) infections, cancer chemotherapy, and anemia in patients who are undergoing surgery. Aranesp is FDA-approved for treatment of anemia resulting from CRF and for use with chemotherapy treatment of nonmyeloid malignancies. The use of ESAs in patients with anemia may decrease the need for and frequency of RBC transfusion.
Pharmacokinetics
ESAs can be administered IV (push) or subQ. According to the manufacturer, ESAs administered by IV are eliminated at a rate consistent with first-order kinetics (process by which the drug is eliminated in part by hepatic and renal blood flow). It has a circulating half-life ranging from approximately 4 to 13 hours in patients with CRF. Plasma levels of Procrit have been detected for at least 24 hours. After subQ administration of Procrit to CRF patients, peak serum levels are achieved within 5 to 24 hours. The half-life of IV-administered Procrit is approximately 20% shorter in normal patients without CRF than in patients with CRF. Prototype Drug Chart 39-1 details the characteristics of this BRM.
 Prototype Drug Chart 39-1
Prototype Drug Chart 39-1 Eprex
Eprex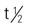 : 4-13 h in patients with CRF; 20% less in those with normal renal function
: 4-13 h in patients with CRF; 20% less in those with normal renal function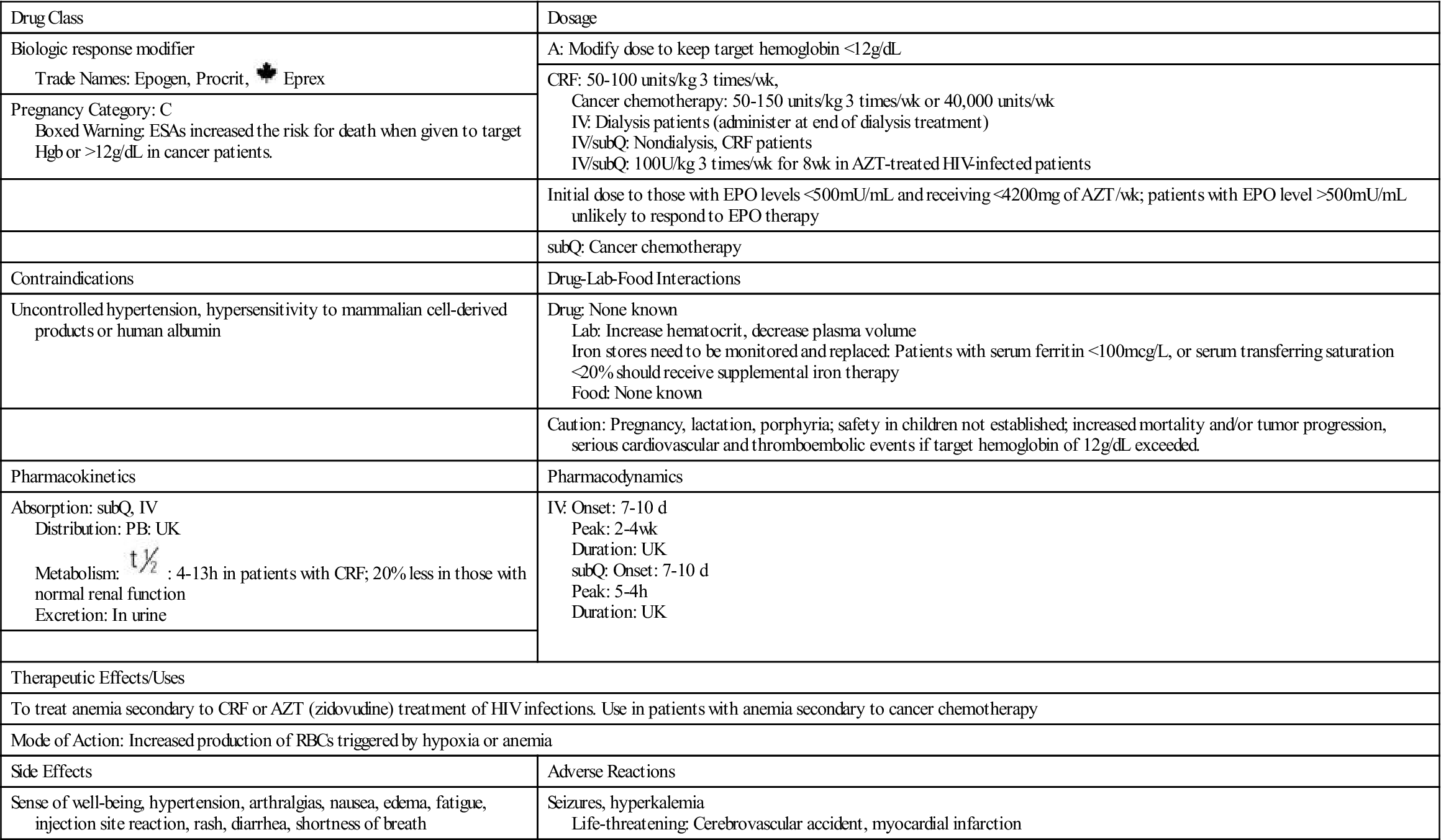
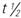 , half-life; UK, unknown; wk, week; <, less than; >, greater than;
, half-life; UK, unknown; wk, week; <, less than; >, greater than;  , Canadian drug name.
, Canadian drug name.Side Effects and Adverse Reactions
It is imperative that the target hemoglobin never exceed 12 g/dL, because the risks of death, serious cardiovascular events, and tumor progression increase at greater values. Close monitoring of hemoglobin response to the ESA and possible dose modification are essential to safe therapy.
The side effects of erythropoietin include hypertension, headache, arthralgias (joint pain), nausea, edema, fatigue, diarrhea, vomiting, chest pain, injection site skin reaction, asthenia (weakness), dizziness, seizures, thrombosis (clots), and allergic reactions. Studies demonstrate that ESA administration is usually well tolerated, and there have been no reports of serious allergic reactions or anaphylaxis. Rare transient skin reactions have been reported. Blood pressure may increase in patients with CRF who receive ESA during the early phase of treatment when the hematocrit (Hct) is on the rise. About 25% of patients with CRF who receive dialysis may require initiation of or increase in antihypertensive therapy. It is postulated that increases in blood pressure may relate to increases in Hct; therefore it is recommended that the dose of ESA be decreased if the Hct increase exceeds 4 points in any 2-week period. Similarly an increase in Hct may cause increased vascular access clotting (clots in the blood vessel at the connection site of the artificial kidney) in hemodialysis patients. Patients may require increased heparinization during ESA therapy to prevent clotting of the artificial kidney. Seizures occurred in patients with CRF who received ESA during clinical trials; activity was particularly evident during the first 90 days of therapy.
Dosing
Procrit should be administered at a starting dose of 50 to 100 units/kg three times a week for patients with CRF. For patients receiving cancer chemotherapy, therapy should not be initiated if the patient’s hemoglobin (Hgb) level is ≥10 g/dL. The Hgb should be monitored on a weekly basis until the level becomes stable. The dose should be titrated for each patient to achieve and maintain the lowest Hgb level sufficient to avoid blood transfusions. The initial recommended dose of Procrit in adults is 150 units/kg 3 times a week or 40,000 units per week given subQ. The dose should be reduced by 25% when the Hgb reaches a level needed to avoid a transfusion or increases >1 g/dL in any 2-week period. The dose should be held if the Hgb exceeds a level needed to avoid transfusion and then restarted at 25% below the previous dose. Aranesp dose is 0.45 mcg/kg subQ every week or 500 mcg subQ every 3 weeks. If the rate of Hgb increase is >1 g/dL in 2 weeks or if the Hgb reaches a level needed to avoid transfusion, the dose should be reduced by 25% of the prior dose. If the Hgb exceeds a level needed to avoid transfusion, the dose should be held until the Hgb approaches a level where transfusion may be required. At this point, the dose should be restarted at 25% of the previous dose. ESAs should be discontinued if after 8 weeks of therapy there is no response as measured by Hgb levels or if transfusions are still required. Otherwise, discontinue ESAs at the completion of the course of chemotherapy.
The following situations must be considered and evaluated if a patient does not respond or maintain a response to an ESA:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


