(sil-DEN-a-fil)
Revatio
Vasodilating agent
Antihypertensive (pulmonary)
Usual dose
2.5 mg (3.125 mL) or 10 mg (12.5 mL) as an IV bolus 3 times daily. Administer doses 4 to 6 hours apart. A 10-mg IV dose of sildenafil is equivalent to a 20-mg oral dose. Resume oral therapy as soon as tolerated. In a study evaluating lower doses, there were no significant differences in the effects on hemodynamic variables compared with higher doses. Treatment with doses higher than 10 mg IV (20 mg PO) three times a day is not recommended.
Dose adjustments
No dose adjustments required for age, gender, race, weight, renal impairment (mild, moderate, or severe), or hepatic impairment (mild or moderate). Has not been studied in patients with severe hepatic impairment.
Dilution
May be given undiluted. Aseptically withdraw the dose directly into a syringe for immediate use.
Filters:
Specific information not available.
Storage:
Store vials at CRT.
Compatibility
Specific information not available. Manufacturer made no recommendations.
Rate of administration
A single dose as an IV bolus.
Actions
An inhibitor of cGMP-specific phosphodiesterase type 5 (PDE-5) in the smooth muscle of the pulmonary vasculature where PDE5 is responsible for degradation of cGMP. Sildenafil increases cGMP within pulmonary vascular smooth muscle cells, resulting in relaxation. In patients with pulmonary hypertension, this can lead to vasodilation of the pulmonary vascular bed and, to a lesser degree, vasodilation in the systemic circulation. PDE5 is also found in other tissues, including vascular and visceral smooth muscle, and in platelets. Sildenafil and its major metabolite are approximately 96% protein bound. Metabolized in the liver predominantly by CYP3A4 and other hepatic microsomal isoenzymes. Terminal half-life is about 4 hours. Findings suggest a lower clearance and/or a higher bioavailability of sildenafil in patients with pulmonary hypertension compared with healthy volunteers. Primarily excreted in feces and approximately 13% in urine.
Indications and uses
Treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) in patients who are currently prescribed oral sildenafil and are temporarily unable to take oral medications. Intended to improve exercise ability and delay clinical worsening. Delay in clinical worsening demonstrated with concurrent use of epoprostenol (Flolan). Studies establishing effectiveness included predominantly patients with NYHA Functional Class II-III symptoms and idiopathic etiology or associated with connective tissue disease.
Limitation of use:
Adding sildenafil to bosentan (Tracleer) therapy in the treatment of PAH does not result in any beneficial effect on exercise capacity.
Contraindications
Known hypersensitivity to sildenafil or any component of the product. ■ Concomitant use of organic nitrates in any form, either regularly or intermittently, because of the greater risk of hypotension. ■ Concomitant use of riociguat (Adempas), a guanylate cyclase stimulator. PDE5 inhibitors, including sildenafil, may potentiate the hypotensive effects of riociguat.
Precautions
Has vasodilatory properties, resulting in mild and transient decreases in BP. Use with caution in patients with resting hypotension (BP less than 90/50 mm Hg), fluid depletion, severe left ventricular outflow obstruction, autonomic dysfunction, and in patients undergoing antihypertensive therapy. ■ Increase in mortality with increasing dose noted in pediatric patients; see Maternal/Child. ■ Not recommended for use in patients with pulmonary veno-occlusive disease (PVOD); it may significantly worsen their cardiac status. Consider the possibility of associated PVOD if signs of pulmonary edema occur. ■ Epistaxis has been reported in patients with PAH secondary to connective tissue disease (CTD) and in patients treated with a concomitant oral vitamin K antagonist (warfarin [Coumadin]). ■ Safety for use in patients with bleeding disorders or active peptic ulceration is unknown. ■ Sudden loss of vision in one or both eyes has been reported. Nonarteritic anterior ischemic optic neuropathy (NAION), a cause of decreased vision (including permanent loss of vision), has been reported in temporal association with PDE-5 inhibitors (sildenafil [Revatio, Viagra], vardenafil [Levitra], tadalafil [Cialis]). Use with caution in patients with retinitis pigmentosa. ■ Sudden decrease or loss of hearing has been reported; may be accompanied by dizziness and tinnitus. ■ Use with caution in patients with an anatomic deformation of the penis (e.g., angulation, cavernosal fibrosis, or Peyronie’s disease) or in patients with conditions that may predispose them to priapism (e.g., sickle cell anemia, multiple myeloma, or leukemia). Penile tissue damage and permanent loss of potency can result from priapism lasting more than 6 hours. ■ Effectiveness and safety of sildenafil in pulmonary hypertension secondary to sickle cell anemia has not been established. Increased incidence of vaso-occlusive crisis requiring hospitalization has been reported in this patient population. ■ See Drug/Lab Interactions.
Monitor:
Obtain baseline studies as indicated by specific patient history. ■ Monitor VS closely to note unsafe drops in BP. Monitoring of BP is especially important when sildenafil is coadministered with other BP-lowering drugs. ■ Monitor for cardiovascular and cerebrovascular events, especially in patients with risk factors. ■ Monitor for vision and/or hearing loss. ■ See Precautions, Patient Education, and Drug/Lab Interactions.
Patient education:
Read manufacturer’s patient education booklet carefully. ■ Never take sildenafil with nitrate medicines or guanylate cyclase stimulators (e.g., riociguat [Adempas]); may cause a sudden and unsafe drop in BP. ■ Do not take Viagra or other similar medications for erectile dysfunction with sildenafil. ■ Seek immediate medical attention with a sudden loss of vision in one or both eyes. ■ Seek prompt medical attention in the event of a sudden decrease or loss of hearing. ■ Seek emergency help if an erection lasts for more than 4 hours. ■ Discuss medications (prescription, nonprescription, and herbal) with a health care professional; see Drug/Lab Interactions.
Maternal/child:
Category B: use during pregnancy only if clearly needed. ■ Use during labor and delivery has not been studied. ■ Not known if sildenafil is secreted in human milk; use caution if required during breast-feeding. ■ Use in pediatric patients, particularly chronic use, is not recommended. In a long-term study, mortality increased; deaths were first observed after about 1 year.
Elderly:
Major differences in response compared with younger adults have not been identified; however, healthy elderly volunteers had a reduced clearance resulting in higher plasma concentrations. Dosing should be cautious. Consider age-related organ impairment and concomitant disease or drug therapy.
Drug/lab interactions
Sildenafil is also marketed as Viagra. Do not take Viagra or other PDE5 inhibitors (e.g., tadalafil [Cialis], vardenafil [Levitra]) concurrently with sildenafil (Revatio). ■ Potentiates the hypotensive effects of nitrates. Concurrent use with nitrates in any form is contraindicated. Nitrates are medicines that treat chest pain (angina) (e.g., nitroglycerin in any form, isosorbide mononitrate [Monoket, Imdur], isosorbide dinitrate [Isordil, Dilatrate-SR], street drugs called “poppers” [amyl nitrate or nitrite]). May result in an unsafe drop in BP. ■ Potentiates the hypotensive effects of riociguat (Adempas), a guanylate cyclase stimulator used to treat pulmonary hypertension. ■ Concurrent use with alpha-adrenergic blocking agents (e.g., alfuzosin [Uroxatral], doxazosin [Cardura], phentolamine, prazosin [Minipress], tamsulosin [Flomax], terazosin [Hytrin]) can lower BP significantly and lead to symptomatic hypotension. BP may be lowered further with this combined use of vasodilators by other variables, including intravascular volume depletion and concomitant use of other antihypertensive drugs (e.g., calcium channel blockers [amlodipine (Norvasc), diltiazem (Cardizem)]). ■ Concomitant use of sildenafil with ritonavir (Norvir) and other potent CYP3A4 inhibitors (e.g., atazanavir [Reyataz], clarithromycin [Biaxin], indinavir [Crixivan], itraconazole [Sporanox], ketoconazole [Nizoral], nefazodone, nelfinavir [Viracept], saquinavir [Invirase], telithromycin [Ketek], and voriconazole [VFEND]) is not recommended. ■ Cimetidine (Tagamet) and erythromycin may cause an increase in sildenafil plasma concentrations. ■ Potent CYP3A4 inducers (e.g., carbamazepine [Tegretol], dexamethasone [Decadron], phenytoin [Dilantin], phenobarbital [Luminal], rifabutin [Mycobutin], rifapentine [Priftin], rifampin [Rifadin], and St. John’s wort) may increase sildenafil clearance and reduce its effectiveness. ■ Bosentan (Tracleer) may also increase sildenafil clearance; see Limitations of Use. ■ Concurrent use with epoprostenol (Flolan) did not have a significant effect on sildenafil pharmacokinetics; effect on epoprostenol not known. ■ Does not potentiate the increase in bleeding time caused by aspirin. ■ Did not potentiate hypotensive effects with alcohol in healthy volunteers. ■ Has been given with atorvastatin [Lipitor], oral contraceptives, and tolbutamide without effect.
Side effects
Manufacturer does not distinguish between side effects caused by oral dosing versus injectable dosing. Most common side effects are dyspepsia, dyspnea, epistaxis, erythema, flushing, headache, insomnia, and rhinitis. Serious side effects include hearing loss, hypotension, indigestion, priapism, vaso-occlusive crisis, and vision loss. Other side effects reported include diarrhea, fever, gastritis, myalgia, paresthesia, and sinusitis. Sildenafil combined with epoprostenol (Flolan) added edema (including peripheral edema), nasal congestion, nausea, and pain in extremities. Retinal hemorrhage occurred in patients with risk factors for hemorrhage, including concurrent anticoagulant therapy. Most hypersensitivity reactions have been nonserious. Serious hypersensitivity reactions (e.g., anaphylaxis, shock) have been rare.
Post-marketing:
Serious cardiovascular, cerebrovascular, and vascular events, including MI, cerebrovascular hemorrhage, hypertension, pulmonary hemorrhage, subarachnoid and intracerebral hemorrhages, TIA, ventricular arrhythmia, and sudden cardiac death; NAION (with decreased vision, including permanent vision loss); sudden decrease or loss of hearing; seizure; and seizure recurrence.
Antidote
Treatment of most side effects will be supportive. Notify physician immediately if a serious side effect occurs (e.g., hearing loss, hypersensitivity, hypotension, priapism, vision loss). Treat hypersensitivity reactions with epinephrine, antihistamines, and corticosteroids as needed. Hemodialysis is not expected to be effective in overdose.
Siltuximab
Sylvant
Monoclonal antibody
Interleukin-6 (IL-6) antagonist
pH 5.2
Usual dose
Pre-testing required:
Absolute neutrophil count (ANC), platelet count, and hemoglobin testing required before each dose for the first 12 months and every 3 dosing cycles thereafter. Treatment criteria are outlined in the following chart. If criteria are not met, consider delaying treatment. Do not reduce dose.
| Siltuximab Treatment Criteria | ||
| Laboratory Parameter | Requirements Before the First Siltuximab Infusion | Retreatment Criteria for Every-3-Week Infusions |
| Absolute neutrophil count | ≥1 × 109/L | ≥1 × 109/L |
| Platelet count | ≥75 × 109/L | ≥50 × 109/L |
| Hemoglobin | <17 Gm/dL | <17 Gm/dL |

Siltuximab:
11 mg/kg as an IV infusion equally distributed over 1 hour. Administer every 3 weeks until treatment failure.
Dose adjustments
No initial dosage adjustment is necessary for patients with CrCl greater than or equal to 15 mL/min. ■ No initial dosage adjustment is necessary for patients with mild to moderate hepatic impairment.
Dilution
Siltuximab is available as a lyophilized powder in 100-mg and 400-mg single-use vials for intravenous infusion. Determine the number of vials required using the following calculations:
Weight in kg × Dose/kg ÷ 100 or 400 mg/vial = # of vials required
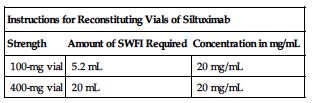
A 60-kg man requiring 11 mg/kg would require 660 mg. 660 mg ÷ 100 mg/vial = 6.6 (÷ 400 mg/vial = 1.65) vials required. After patient is weighed and appropriate dose is calculated, remove vials from the refrigerator and allow to come to RT for 30 minutes. Use of a 21-gauge 11/2-inch needle is recommended for preparation. Aseptically reconstitute each siltuximab vial as instructed in the following chart.
| Instructions for Reconstituting Vials of Siltuximab | ||
| Strength | Amount of SWFI Required | Concentration in mg/mL |
| 100-mg vial | 5.2 mL | 20 mg/mL |
| 400-mg vial | 20 mL | 20 mg/mL |
Gently swirl reconstituted vials. Do Not Shake or Swirl Vigorously. Powder may take up to 60 minutes to dissolve. Must be further diluted for infusion within 2 hours of the initial reconstitution. Maintain at RT. Do not use if particles or solution discoloration is present or if solution is visibly opaque. To further dilute, withdraw a volume of D5W equal to the calculated volume of the siltuximab dose from a 250-mL infusion bag of D5W and slowly add the siltuximab solution. Gently invert to mix the solution. Infusion bags must be made of either polyvinyl chloride (PVC) with di(2-ethylhexyl) phthalate (DEHP) or polyolefin (PO).
Filters:
Use of a 0.2-micron inline polyethersulfone (PES) filter required.
Storage:
Store unopened vials at 2° to 8° C (36° to 46° F) in original cartons to protect from light. Do not use beyond expiration date located on the carton and vial. Complete infusion within 4 hours of dilution. Discard any unused portion of the reconstituted product or the infusion solution. Waste material should be disposed of in accordance with local requirements.
Compatibility
Manufacturer states, “Do not infuse siltuximab concomitantly in the same intravenous line with other agents.” Compatible only with infusion bags of polyvinyl chloride (PVC) with di(2-ethylhexyl) phthalate (DEHP) or polyolefin (PO) and administration sets lined with PVC, DEHP, or polyurethane (PU).
Rate of administration
A single dose equally distributed over 1 hour as an IV infusion. Stop the infusion if the patient develops a mild to moderate infusion reaction. If the reaction resolves, siltuximab may be restarted at a lower infusion rate. Consider premedication with antihistamines, acetaminophen, and corticosteroids as indicated. Discontinue siltuximab if patient does not tolerate the infusion following these interventions. Complete infusion within 4 hours of dilution.
Actions
Siltuximab is a human-mouse chimeric monoclonal antibody produced by Chinese hamster ovary cells. It binds human interleukin-6 (IL-6) and prevents the binding of IL-6 to both soluble and membrane-bound IL-6 receptors. Overproduction of IL-6 has been linked to systemic manifestations in patients with multicentric Castleman’s disease (MCD). With the first infusion, peak serum concentration occurs close to the end of the infusion. With the once-every-3-week dosing regimen, siltuximab steady-state is achieved by the sixth infusion. The mean terminal half-life for siltuximab in patients after the first infusion is 20.6 days (range 14.2 to 29.7 days).
Indications and uses
Treatment of patients with multicentric Castleman’s disease (MCD) who are human immunodeficiency virus (HIV) negative and human herpesvirus-8 (HHV-8) negative.
Limitation of use:
Siltuximab was not studied in patients with MCD who are HIV-positive or HHV-8 positive because siltuximab did not bind to virally produced IL-6 in a nonclinical study.
Contraindications
Severe hypersensitivity reaction to siltuximab or any of its excipients.
Precautions
Administered under the direction of a physician knowledgeable in its use in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Do not administer siltuximab to patients with severe infections until the infection resolves. May mask signs and symptoms of acute inflammation, including suppression of fever and of acute-phase reactants such as C-reactive protein (CRP). ■ Hypersensitivity and/or infusion reactions, including anaphylaxis, may occur. ■ Do not administer live virus vaccines to patients receiving siltuximab. IL-6 inhibition may interfere with the normal immune response to new antigens. ■ Use with caution in patients who may be at increased risk for GI perforation. ■ May increase hemoglobin. ■ Effects of severe renal impairment (CrCl less than 15 mL/min) on siltuximab pharmacokinetics have not been determined. ■ Patients with severe hepatic impairment (Child-Pugh Class C) were not included in clinical trials. ■ A protein substance, it has the potential for producing an immune response. Anti-siltuximab antibodies have been detected in a small number of patients; clinical significance unknown.
Monitor:
Obtain ANC, platelets, and hemoglobin before each dose for the first 12 months and every 3 dosing cycles thereafter. Consider delay of treatment if established parameters are not met. Do not reduce dose; see Usual Dose. ■ Monitor closely for S/S of infections. Prompt anti-infective therapy is recommended. Do not administer further siltuximab until the infection resolves. ■ Monitor for S/S of a hypersensitivity reaction (e.g., chest pain, chills, dizziness, dyspnea, fever, flushing, hypotension, nausea, pruritus, rash, urticaria) or infusion reaction (e.g., back pain, chest pain or discomfort, erythema, flushing, palpitations, nausea and vomiting). ■ Promptly evaluate patients presenting with symptoms that may be associated or suggestive of GI perforation. ■ Monitor uric acid levels as indicated. May be treated with adequate hydration and, if necessary, with allopurinol and alkalinization of urine. ■ Assess patient’s overall health at each treatment visit.
Patient education:
Review manufacturer’s medication guide before each dose. ■ Discuss recommended vaccinations before treatment with siltuximab. ■ Promptly report S/S of serious hypersensitivity or infusion reaction during the infusion. S/S include chest tightness, difficulty breathing, severe dizziness or light-headedness, swelling of the lips, skin rash, and wheezing. ■ May lower resistance to infections; promptly report S/S of infection (e.g., fever, sore throat, unusual redness around a sore). ■ Report any S/S of new or worsening medical conditions. ■ Avoid pregnancy. Women of childbearing potential should use contraception during treatment and for 3 months after treatment.
Maternal/child:
Category C: avoid pregnancy; use during pregnancy only if potential benefit justifies the risk to the fetus. ■ Use of contraception indicated during and for 3 months after treatment is complete. ■ Infants born to pregnant women taking siltuximab may be at an increased risk for infection. Caution is advised in the administration of live virus vaccines to these infants. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
Differences in response compared with younger adults have not been identified, but greater sensitivity of older individuals cannot be ruled out.
Drug/lab interactions
No in vitro or in vivo drug-drug interaction studies have been conducted with siltuximab. ■ Inhibition of IL-6 signaling in patients treated with siltuximab may increase metabolism of CYP450 substrates. When initiating or discontinuing siltuximab in patients being treated with CYP450 substrates with a narrow therapeutic index, monitoring of effect (e.g., INR with warfarin [Coumadin]) or drug concentrations (e.g., cyclosporine [Sandimmune], theophylline) may be required. Adjust dose as needed. The effect of siltuximab on CYP450 enzyme activity can persist for several weeks after stopping therapy. Use caution when siltuximab is coadministered with CYP3A4 substrates when a decrease in effectiveness would be undesirable (e.g., atorvastatin [Lipitor], lovastatin [Mevacor], oral contraceptives). ■ Do not administer live virus vaccines to patients receiving siltuximab.
Side effects
The most commonly reported side effects were hyperuricemia, increased weight, pruritus, rash (rash generalized, rash maculopapular, rash popular, and rash pruritic), and upper respiratory tract infection. Concurrent active serious infections as well as hypersensitivity and/or infusion-related reactions may occur. Other side effects reported include arthralgia, constipation, diarrhea, dry skin, eczema, edema (generalized, localized), fatigue, headache, hypercholesterolemia, hypertriglyceridemia, hypotension, lower respiratory tract infection, oropharyngeal pain, pain in extremities, psoriasis, renal impairment, and thrombocytopenia.
Antidote
Keep physician informed of side effects; may be treated symptomatically if indicated. Based on the severity of the reaction, slow the infusion rate, temporarily interrupt, or discontinue the infusion. Manage acute reactions that require intervention by either temporarily interrupting or discontinuing the infusion and administering antihistamines, antipyretics, or corticosteroids as necessary. Discontinue siltuximab in patients with severe hypersensitivity and/or infusion-related reactions, anaphylaxis, or cytokine release syndromes. Do not reinstitute treatment. Treat with oxygen, epinephrine (Adrenalin), antihistamines (e.g., diphenhydramine [Benadryl]), vasopressors (e.g., dopamine), corticosteroids, albuterol, IV fluids, and ventilation equipment as indicated. Resuscitate as necessary.
Sipuleucel-T
Provenge
Autologous cellular immunotherapy
Antineoplastic
Usual dose
An autologous (derived from the same individual) cellular immunotherapy. Each infusion of sipuleucel-T is preceded by a leukapheresis procedure and a specific preparation procedure by the manufacturer; see Dilution and Actions. Three complete doses are administered at approximately 2-week intervals (range of 1 to 15 weeks in clinical trials).
Approximately 3 days before the desired infusion date:
Leukapheresis:
Peripheral blood mononuclear cells are obtained via a standard leukapheresis procedure. These cells are then sent to the manufacturer (Dendreon) to be prepared for reinfusion into the patient.
Day of infusion:
Premedication:
Administer oral acetaminophen and an antihistamine (e.g., diphenhydramine [Benadryl]) 30 minutes before administration to minimize potential acute infusion reactions (e.g., chills, fever).
Sipuleucel-T:
For autologous IV use only. Do not use a cell filter. Confirm that the patient’s identity matches the patient identifiers on the infusion bag and the Cell Product Disposition Form (CPDF). Each dose contains a minimum of 50,000,000 autologous CD54+ cells activated with PAP-GM-CSF and suspended in 250 mL of LR in a sealed, patient-specific infusion bag. Do not infuse until confirmation of product release has been received from Dendreon; see Dilution. Infusion must begin before the expiration date and time on the Cell Product Disposition Form (CPDF).
Dilution
Will be shipped directly to the infusing provider in packaging intended to protect the infusion bag and maintain storage temperatures until infusion. Verify the product and patient-specific labels located on top of the insulated container. Do not remove from the shipping box or open the lid of the insulated container until the patient is ready for infusion. The manufacturer will send to the infusion site a CPDF that contains the patient identifiers, expiration date and time, and disposition status (approved for infusion or rejected). When all preparations have been made for infusion and the CPDF has been received and verified, remove the sipuleucel-T infusion bag from the insulated container and inspect for signs of leakage. Contents will be slightly cloudy and a cream-to-pink color. Gently mix and resuspend contents, inspecting for clumps and clots. Small clumps of cellular material should disperse with gentle manual mixing. Do not administer if the bag leaks during handling or if clumps remain in the bag.
Filters:
Do not use a cell filter.
Storage:
After removal from the insulated container, the infusion must be complete within 3 hours at RT. Do not return to the shipping container. Do not initiate infusion of expired sipuleucel-T.
Compatibility
Specific information not available; however, specific use indicates it should be administered separately; consult pharmacist.
Rate of administration
A single dose of 250 mL as an infusion equally distributed over 1 hour. Do not use a cell filter. If an infusion reaction occurs, decrease rate or interrupt infusion depending on the severity of the reaction; see Antidote.
Actions
An autologous cellular immunotherapy. The patient’s own immune cells are obtained through leukapheresis. Active components of sipuleucel-T are autologous peripheral blood mononuclear cells, including antigen-presenting cells (APCs) and PAP-GM-CSF (prostatic acid phosphatase [PAP], an antigen expressed in prostate cancer tissue, linked to granulocyte-macrophage colony-stimulating factor [GM-CSF], an immune cell activator). The recombinant antigen can bind to and be processed by APCs into smaller protein fragments. The recombinant antigen is designed to target APCs and may help direct the immune response to PAP. The final product contains T-cells, B cells, natural killer (NK) cells and other cells.
Indications and uses
Treatment of asymptomatic or minimally symptomatic metastatic hormone refractory prostate cancer.
Contraindications
Manufacturer states, “None.”
Precautions
Intended solely for autologous use. For IV use only. ■ Administered under the direction of a physician knowledgeable in its use in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Concurrent use of immunosuppressive agents may alter the effectiveness and/or safety of sipuleucel-T; see Drug/Lab Interactions. ■ Health care professionals must use universal precautions when handling leukapheresis material and sipuleucel-T. Neither is routinely tested for transmissible infectious diseases and thus may carry the risk of infectious disease transmission to health professionals during handling of the products. ■ Acute infusion reactions (reported within 1 day of infusion) occur frequently and range from mild to serious; see Monitor. Incidence increased with the second infusion and decreased after the third infusion.
Monitor:
Obtain baseline vital signs and repeat as indicated. ■ Observe the patient for S/S of an acute infusion reaction (e.g., bronchospasm, chills, fatigue, fever, hypertension, joint aches, nausea, tachycardia) during the infusion and for at least 30 minutes after the infusion. If an acute infusion reaction occurs, the infusion may be interrupted or slowed depending on the severity of the reaction. Do not resume the infusion if the bag will be held at RT for more than 3 hours. ■ Closely monitor patients with cardiac or pulmonary conditions. ■ If the patient is unable to receive a scheduled infusion, the patient will need to undergo an additional leukapheresis procedure. ■ Monitor for extravasation.
Patient education:
Maintain all scheduled appointments. ■ An additional leukapheresis procedure will be required if a scheduled infusion cannot be completed. ■ A central venous line may be required if peripheral venous access is not adequate to accommodate the leukapheresis procedure and/or the infusion. ■ Promptly report S/S of an infusion reaction (e.g., breathing problems, chills, dizziness, fatigue, fever, headache, hypertension, muscle aches, nausea, or vomiting). ■ Report S/S that may indicate a cardiac arrhythmia (e.g., very slow or rapid pulse). ■ Review prescription and nonprescription drug profile with a physician; see Drug/Lab Interactions.
Maternal/child:
Not indicated for use in these populations.
Elderly:
Safety similar to that seen in younger adults.
Drug/lab interactions
Drug interaction studies have not been performed. ■ Use of either chemotherapy or immunosuppressive agents (such as systemic corticosteroids) given concurrently with the leukapheresis procedure or sipuleucel-T has not been studied. ■ Sipuleucel-T is designed to stimulate the immune system, and concurrent use of immunosuppressive agents may alter the effectiveness and/or safety of sipuleucel-T. Evaluate patients carefully for the medical appropriateness of reducing or discontinuing immunosuppressive agents before treatment.
Side effects
The most common events of backache, chills, fatigue, fever, headache, joint aches, and nausea are usually mild or moderate and generally resolve within 2 days. Other side effects included anemia, anorexia, citrate toxicity, constipation, cough, diarrhea, hematuria, hot flush, influenza-like illness, insomnia, muscle aches and spasms, pain (back, bone, chest [musculoskeletal], extremity, and neck), paresthesia (oral, extremity), peripheral edema, rash, sweating, tremor, URIs, UTIs, weight loss. Acute infusion reactions (mild to moderate) occur frequently. Severe acute infusion reactions (Grade 3) add asthenia, bronchospasm, dizziness, dyspnea, hypertension, hypoxia, and vomiting. Cerebrovascular events (e.g., hemorrhagic and ischemic strokes) and single reports of eosinophilia, myasthenia gravis, myositis, rhabdomyolysis, and tumor flare have been reported.
Antidote
Keep the physician informed of significant side effects and treat as appropriate. For acute infusion reactions, the infusion may be interrupted or slowed depending on the severity of the reaction. Do not resume the infusion if the bag will be held at RT for more than 3 hours. In controlled clinical trials, acute infusion reactions were treated with acetaminophen, IV antihistamines (e.g., diphenhydramine [Benadryl]) and/or H2 blockers (e.g., ranitidine [Zantac]) and low-dose IV meperidine. Resuscitate as indicated.
Sodium acetate
(SO-dee-um AS-ah-tayt)
Electrolyte replenisher
Antihyponatremic
Alkalizing agent
pH 6 to 7
Usual dose
Determined by nutritional needs, evaluation of electrolytes, and degree of hyponatremia. Some solutions may contain aluminum; see Precautions. Available in 2 mEq and 4 mEq/mL concentrations. Each mL provides 2 or 4 mEq each of sodium and acetate.
Dose adjustments
Lower-end initial doses may be appropriate in the elderly based on the potential for decreased organ function and concomitant disease or drug therapy.
Dilution
Must be added to larger volumes of IV infusion solutions including total parenteral nutrition. Use only clear solutions. Check labels for aluminum content; see Precautions.
Storage:
Store at room temperature. Discard unused portion.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Y-site:
Enalaprilat (Vasotec IV), esmolol (Brevibloc), labetalol, nicardipine (Cardene IV), ondansetron (Zofran).
Rate of administration
Administer at prescribed rate for infusion solutions. Rapid or excessive administration may produce sodium overload, water retention, alkalosis, or hypokalemia.
Actions
An alkalizing agent and sodium salt. Sodium is the predominant cation of extracellular fluid. It controls water distribution throughout the body. Hypothalamus osmoreceptors, sensitive to osmolarity changes in the blood, control serum sodium concentration (142 mEq/L). Body fluid is lost when sodium content decreases and retained when sodium content increases. Sodium is excreted by the kidney. The acetate ion is metabolized to bicarbonate, thus providing a source of bicarbonate. It also acts as a hydrogen ion receptor.
Indications and uses
To prevent or correct hyponatremia in patients with restricted intake, especially in individualized IV formulations when basic needs are not met by standard solutions. ■ Treatment of mild to moderate acidotic states. ■ Source of sodium ions in hemodialysis and peritoneal dialysis.
Contraindications
Patients with hypernatremia or water retention.
Precautions
Use with caution in impaired renal function, congestive heart failure, hypertension, peripheral or pulmonary edema, any condition resulting in salt retention, and in patients receiving corticosteroids. ■ Use acetate-containing solutions with extreme caution in patients with metabolic or respiratory alkalosis and/or impaired hepatic function. ■ Temporary therapy in acidosis. Treatment of primary condition must be instituted. ■ Sodium bicarbonate is the drug of choice for use in severe acidosis that requires immediate correction. ■ Some solutions of sodium acetate contain aluminum. In impaired kidney function, aluminum may reach toxic levels. Premature neonates are particularly at risk because of their immature kidneys and requirement for calcium and phosphate, which also contain aluminum. Research indicates that patients with impaired renal function who receive more than 4 to 5 mcg/kg/day of parenteral aluminum are at risk for developing CNS or bone toxicity associated with aluminum accumulation.
Monitor:
Evaluate electrolytes frequently during treatment. ■ Evaluate fluid balance. ■ Rapid or excessive administration may produce alkalosis or hypokalemia. Cardiac arrhythmias may result from an intracellular shift of potassium. Many other complications may arise from electrolyte imbalance.
Maternal/child:
Category C: safety not established; use only if clearly needed.
Elderly:
Differences in response between elderly and younger patients have not been identified. Lower-end initial doses may be appropriate in the elderly; see Dose Adjustments.
Drug/lab interactions
Alkalinization of urine may increase the renal elimination and decrease the effects of many drugs, including tetracyclines, chlorpropamide, lithium carbonate, methotrexate, salicylates, and may decrease the renal elimination and prolong the effects of others, including anorexiants (e.g., amphetamines), flecainide, mecamylamine, quinidine, sympathomimetics (e.g., dopamine, ephedrine).
Side effects
Hypernatremia, sodium level over 147 mEq/L, is most common (congestive heart failure, delirium, dizziness, edema, fever, flushing, headache, hypotension, oliguria, pulmonary edema, reduced salivation and lacrimation, respiratory arrest, restlessness, swollen tongue, tachycardia, thirst, weakness). Alkalosis and fluid or solute overload can occur.
Antidote
Notify the physician of any side effect. Reduce rate and notify physician at first sign of congestion or fluid overload. May be treated by sodium restriction and/or use of diuretics (e.g., furosemide [Lasix]) or dialysis. Resuscitate as necessary.
Sodium bicarbonate
(SO-dee-um bye-KAR-bon-ayt)
Electrolyte replenisher
Alkalizing agent
pH 7 to 8.5
Usual dose
Adjusted according to pH, Paco2, calculated base deficit, clinical response, and fluid limitations of the patient. In the presence of a low CO2 content, adjust gradually to avoid unrecognized alkalosis. Correction to a CO2 of 20 mEq/L within 24 hours will most likely result in a normal pH if the cause of acidosis is controlled and normal kidney function is present. Average dose for most indications is 2 to 5 mEq/kg/24 hr in adults and pediatric patients.
Cardiac arrest:
1 mEq/kg of body weight, only when appropriate (see Precautions; evidence supports little benefit and use may be detrimental). Repeat half dose in 10 minutes if indicated by blood pH and Paco2.
Pediatric dose
0.5 to 1 mEq/kg. For neonates and children up to 2 years of age, dose must never exceed 8 mEq/kg/24 hr of a 4.2% or more dilute solution; see Usual Dose, Monitor, and Maternal/Child.
Dilution
4.2% sodium bicarbonate solution:
5 mEq/10 mL (0.5 mEq/mL).
5% sodium bicarbonate solution:
297.5 mEq/500 mL (0.595 mEq/mL).
7.5% sodium bicarbonate solution:
44.6 mEq/50 mL (0.892 mEq/mL).
8.4% sodium bicarbonate solution:
50 mEq/50 mL (1 mEq/mL).
Neut (4% sodium bicarbonate solution):
2.4 mEq/5 mL (0.48 mEq/mL). Use limited to a buffering solution. Will raise pH of IV fluids and medications. Never used as a systemic alkalinizer.
May be given in prepared solutions. 7.5% and 8.4% solutions should be diluted with equal amount of water for injection, or diluted with compatible IV solutions, depending on desired dosage and desired rate of administration. 4.2% or a more dilute solution is preferred for infants and children. Use only clear solutions.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Will form a precipitate with many drugs, including epinephrine. If coadministration with epinephrine is indicated, give at separate sites.
One source suggests the following compatibilities:
Additive:
Amikacin, aminophylline, amphotericin B (conventional), atropine, calcium chloride, cefoxitin (Mefoxin), ceftazidime (Fortaz), ceftriaxone (Rocephin), chloramphenicol (Chloromycetin), clindamycin (Cleocin), cytarabine (ARA-C), erythromycin (Erythrocin), esmolol (Brevibloc), furosemide (Lasix), heparin, levofloxacin (Levaquin), lidocaine, magnesium sulfate, mannitol, meperidine (Demerol), methotrexate, methyldopate, multivitamins (M.V.I.), nafcillin (Nallpen), oxytocin (Pitocin), penicillin G potassium, phenylephrine (Neo-Synephrine), phytonadione (vitamin K1), potassium chloride (KCl), prochlorperazine (Compazine), verapamil.
Y-site:
Acyclovir (Zovirax), amifostine (Ethyol), aztreonam (Azactam), bivalirudin (Angiomax), ceftaroline (Teflaro), ceftriaxone (Rocephin), ciprofloxacin (Cipro IV), cisatracurium (Nimbex), cladribine (Leustatin), cyclophosphamide (Cytoxan), cytarabine (ARA-C), daunorubicin (Cerubidine), dexamethasone (Decadron), dexmedetomidine (Precedex), diltiazem (Cardizem), docetaxel (Taxotere), doripenem (Doribax), doxorubicin (Adriamycin), etoposide (VePesid), etoposide phosphate (Etopophos), famotidine (Pepcid IV), filgrastim (Neupogen), fludarabine (Fludara), gallium nitrate (Ganite), gemcitabine (Gemzar), granisetron (Kytril), heparin, hydrocortisone sodium succinate (Solu-Cortef), 6% hydroxyethyl starch (Voluven), ifosfamide (Ifex), indomethacin (Indocin IV), insulin (regular), levofloxacin (Levaquin), linezolid (Zyvox), melphalan (Alkeran), mesna (Mesnex), methylprednisolone (Solu-Medrol), milrinone (Primacor), morphine, paclitaxel (Taxol), pemetrexed (Alimta), piperacillin/tazobactam (Zosyn), potassium chloride (KCl), propofol (Diprivan), remifentanil (Ultiva), tacrolimus (Prograf), telavancin (Vibativ), teniposide (Vumon), thiotepa, vancomycin, vasopressin.
Rate of administration
Flush IV line thoroughly before and after administration. Usual rate of administration of any solution is 2 to 5 mEq/kg over 4 to 8 hours. Do not exceed 50 mEq/hr. Decrease rate for pediatric patients. See Pediatric Dose. Rapid or excessive administration may produce alkalosis, hypernatremia, hypocalcemia, and hypokalemia. Cardiac arrhythmias may result from an intracellular shift of potassium. Will also produce pain and irritation along injection site.
Cardiac arrest:
Up to 1 mEq/kg properly diluted over 1 to 3 minutes.
Actions
An alkalizing agent and sodium salt. Helps to maintain osmotic pressure and ion balance. It is the buffering agent in blood. Bicarbonate ion elevates blood pH promptly. 99% reabsorbed with normal kidney function. Only 1% is excreted in the urine.
Indications and uses
Metabolic acidosis (blood pH below 7.2 or plasma bicarbonate of 8 mEq/L or less) caused by circulatory insufficiency resulting from shock or severe dehydration, extracorporeal circulation of blood, severe renal disease, cardiac arrest (see Precautions), uncontrolled diabetes with ketoacidosis (low-dose insulin preferred), and primary lactic acidosis. ■ Hyperkalemia. ■ Hemolytic reactions requiring alkalinization of urine to reduce nephrotoxicity. ■ Severe diarrhea. ■ Barbiturate, methyl alcohol, or salicylate intoxication. ■ Buffering solution to raise pH of IV fluids and medications.
Contraindications
Diuretics known to produce hypochloremic alkalosis (e.g., thiazides), edema, hypertension, hypocalcemia (alkalosis may produce CHF, convulsions, hypertension, and tetany), hypochloremia (from vomiting, GI suction, or diuretics), hypernatremia, impaired renal function, metabolic alkalosis, respiratory alkalosis or acidosis, and any situation in which the administration of sodium could be clinically detrimental.
Precautions
Temporary therapy in metabolic acidosis. Treatment of primary condition must be instituted. Best to partially correct acidosis and allow compensatory mechanisms to complete the correction. ■ Use with caution in cardiac, liver, or renal disease; CHF; fluid/solute overload; elderly and postoperative patients with renal or cardiovascular insufficiency; and in patients receiving corticosteroids. ■ Use in cardiac arrest indicated only in cases of prolonged resuscitation with effective ventilation or after return of spontaneous circulation after a long arrest interval. ■ Adequate alveolar ventilation should control acid-base balance in most arrest situations except prolonged cardiac arrest, arrested patients with pre-existing metabolic acidosis, hyperkalemia, or tricyclic or barbiturate overdose.
Monitor:
Confirm absolute patency of vein. Extravasation may cause chemical cellulitis, necrosis, ulceration, or sloughing. ■ Flush IV line thoroughly before and after administration; many incompatibilities. ■ Determine blood pH, Po2, Pco2, and electrolytes several times daily during intensive treatment and daily in most other situations. Determine base excess or deficit in infants and children (dose = 0.3 × kg × base deficit). Notify physician of all results. ■ Rapid or excessive administration may produce alkalosis, hypokalemia, and hypocalcemia. Cardiac arrhythmias may result from an intracellular shift of potassium. Many other complications may arise from electrolyte imbalance. ■ Use only 50-mL ampules in cardiac arrest to prevent accidental overdose. Recent practice indicates smaller doses may be appropriate when indicated in cardiac arrest and may prevent secondary alkalosis. Adequate alveolar ventilation is imperative. Evaluate patient response and blood gases.
Maternal/child:
Category C: safety for use in pregnancy not established; use only if clearly needed. ■ Use caution in breast-feeding. ■ Doses in excess of 8 mEq/kg/24 hr and/or given too rapidly (10 mL/min) may cause intracranial hemorrhage, hypernatremia, and decrease in cerebrospinal fluid pressure in neonates and children under 2 years.
Elderly:
Contains sodium; use caution in the elderly with renal or cardiovascular insufficiency with or without CHF; see Precautions.
Drug/lab interactions
Alkalinization of urine may increase the renal elimination and decrease the effects of many drugs, including tetracyclines, chlorpropamide, lithium carbonate, methotrexate, salicylates, and may decrease the renal elimination and prolong the effects of others, including anorexiants (e.g., amphetamines), flecainide, mecamylamine, quinidine, sympathomimetics (e.g., dopamine, ephedrine).
Side effects
Rare when used with caution: alkalosis (hyperirritability and tetany), hypernatremia (edema, CHF), hypokalemia, local site venous irritation.
Antidote
Discontinue the drug and notify the physician of any side effect. Hypokalemia usually occurs with alkalosis. Sodium and potassium chloride must be supplemented as indicated for correction. Treatment of alkalosis often results in more alkalosis. Rebreathing expired air from a paper bag may help to control beginning symptoms of alkalosis. Calcium gluconate may help in severe alkalosis. Administration of a balanced hypotonic electrolyte solution (Isolyte H, Normosol-M, Plasma-lyte 56) with sodium and potassium chloride added may help to excrete the bicarbonate ion in the urine. Ammonium chloride may be indicated. Treat tetany as indicated (calcium gluconate). For extravasation, discontinue infusion; aspirate fluid, drug, and/or 3 to 5 mL of blood through the in-place needle, then remove the needle. Elevate the extremity and apply warm, moist compresses. Resuscitate as necessary.
Sodium chloride
(SO-dee-um KLOR-eyed)
Electrolyte replenisher
Antihyponatremic
pH 4.5 to 7
Usual dose
Highly individualized and dependent on age, weight, clinical condition of patient, concentration of salts in the plasma, and/or loss of body fluids.
Hypotonic:
(0.45% [1/2NS], 4.5 Gm of sodium chloride/L or 77 mEq of sodium and 77 mEq of chloride [approximately 155 mOsm/L]) 2 to 4 L/24 hr.
Isotonic:
(0.9% [NS], 9 Gm of sodium chloride/L or 154 mEq of sodium and 154 mEq of chloride [approximately 310 mOsm/L]) 1.5 to 3 L/24 hr.
Bacteriostatic isotonic NS contains benzyl alcohol as a preservative. It is used in small amounts (usually 1 to 2 mL) as a diluent for injectable drugs (IV, IM, SC) or to flush IV lines. One study suggests that up to 30 mL/dose may be used in adults. However, the amount of benzyl alcohol that is tolerated within 24 hours in adults without toxic effects has not been determined. Use only preservative-free sodium chloride in newborns for all indications.
Hypertonic:
Calculate sodium deficit. Total body water (TBW) is 45% to 50% in females and 50% to 60% in males.
Na deficit in mEq = TBW [desired − observed plasma Na]
Hypertonic (3%, 30 Gm of sodium chloride/L or 513 mEq of sodium and 513 mEq of chloride [approximately 1,030 mOsm/L] or 5%, 50 Gm of sodium chloride/L or 855 mEq of sodium and 855 mEq of chloride [approximately 1,710 mOsm/L]), 200 to 400 mL/24 hr. To correct acute serious hyponatremia, hypertonic sodium chloride is used to correct the serum sodium in 5 mEq/L/dose increments at a rate of no more than 0.5 mEq/hr until serum sodium is 125 mEq/L or neurologic symptoms improve. In the first 3 to 4 hours, an increase of plasma sodium at rates up to 1 mEq/L/hr may be tolerated in patients with distressing symptoms. To prevent an overly rapid correction, an increase in plasma sodium of less than 10 mEq/L in the first 24 hours and an increase of less than 18 mEq/L in the first 48 hours is desired. See Precautions.
Concentrated:
To be used only as an additive in parenteral fluid therapy (14.6% contains 2.5 mEq of sodium and chloride/mL; 23.4% contains 4 mEq of sodium and chloride/mL).
Dose adjustments
Dose selection should be cautious in the elderly, especially with hypertonic or concentrated solutions. Start at the lower end of the desired dosing range; consider the greater frequency of decreased organ function and of concomitant disease or drug therapy.
Dilution
Available as hypotonic 25 mL, 50 mL, 150 mL, 250 mL, 500 mL, 1 L; isotonic (2 mL, 3 mL, 5 mL, 10 mL, 20 mL, 25 mL, 30 mL, 50 mL, 100 mL, 150 mL, 250 mL, 500 mL, 1 L); or hypertonic (500 mL) solution in vials and/or bottles for injection or infusion and ready for use. Isotonic and hypotonic sodium chloride are frequently combined with 5% or 10% dextrose. Concentrated must be diluted before use. Used only as an additive in parenteral fluids. Permits specific mEq for mEq replacement of sodium and chloride without contributing to fluid overload. Available in 14.6% strength in 20 mL, 40 mL, and 200 mL; 23.4% strength in 30 mL, 50 mL, 100 mL, and 200 mL.
Bacteriostatic isotonic available in 2-mL, 10-mL, and 30-mL vials ready for use as a diluent. Never use in neonates.
Storage:
Store at CRT. Do not freeze.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibility for concentrations of 14.6% and 23.4%:
Y-site:
Ciprofloxacin (Cipro IV).
Rate of administration
IV infusions in flexible plastic containers:
Do not hang in series connection, do not pressurize to increase flow rates without first fully evacuating residual air from the container, and do not use vented IV administration sets. All may result in air embolism.
Isotonic and hypotonic:
A single daily dose equally distributed over 24 hours. Rate is dependent on age, weight, and clinical condition of the patient.
Hypertonic:
One-half the calculated dose over at least 8 hours. Do not exceed 100 mL over 1 hour. Too-rapid infusion may cause local pain and venous irritation; reduce rate for tolerance; see Usual Dose.
Concentrated:
Properly diluted in parenteral fluids and equally distributed over 24 hours. Never exceed hypertonic rate (see above) based on actual mEq of sodium chloride.
Actions
Sodium:
The predominant cation of extracellular fluid. It controls water distribution throughout the body. Hypothalamus osmoreceptors, sensitive to osmolarity changes in the blood, control serum sodium concentration (142 mEq/L). Body fluid is lost when sodium content decreases and retained when sodium content increases.
Chloride:
The major extracellular anion. Closely follows the metabolism of sodium. Changes in the acid-base balance of the body are reflected by the changes in chloride concentration.
Distribution and excretion of sodium and chloride are largely under control of the kidney, which maintains a balance between intake and output.
Indications and uses
Replace lost fluid or sodium and chloride ions in the body (e.g., hyponatremia, low salt syndrome, dehydration). ■ Maintain fluid and electrolyte balance.
Hypotonic:
Water replacement without increase of osmotic pressure or serum sodium levels; treatment of hyperosmolar diabetes requiring considerable fluid without excess sodium.
Isotonic:
To replace sodium and chloride lost from vomiting because of obstructions and/or aspiration of GI fluids; treatment of metabolic alkalosis with fluid loss and sodium depletion. ■ Diluent in parenteral preparations. ■ To initiate and terminate blood transfusions without hemolyzing RBCs. ■ Maintain patency and perform routine irrigations of many types of intravascular devices (e.g., catheters, implanted ports). ■ Antidote for drug-induced hypercalcemia. Given concurrently with furosemide (Lasix). ■ Priming solution in hemodialysis procedures.
Hypertonic:
Used only when high sodium and/or chloride content without large amounts of fluid is required (e.g., electrolyte and fluid loss replaced with sodium-free fluids, excessive water intake resulting in drastic dilution of body water, emergency treatment of severe salt depletion, addisonian crisis, diabetic coma).
Concentrated:
Used to meet the specific requirements of patients with unusual fluid and electrolyte needs (e.g., special problems of sodium electrolyte intake or excretion).
Contraindications
Hypernatremia; fluid retention; situations where sodium or chloride could be detrimental. 3% and 5% sodium chloride solutions are contraindicated with elevated, normal, or slightly decreased serum sodium and chloride levels. Bacteriostatic sodium chloride is contraindicated in newborns.
Precautions
Use caution in circulatory insufficiency, congestive heart failure, edema with sodium retention, kidney dysfunction, hepatic disease, hypoproteinemia, in the elderly or debilitated individuals, and in patients receiving corticosteroids. ■ Use with caution in surgical patients; see Monitor. ■ More than 1 liter of NS may cause hypernatremia, which can result in loss of bicarbonate ions and acidosis. ■ All uses require preservative-free solutions except the limited use of bacteriostatic NS as a diluent or flushing agent. ■ Inadvertent direct injection or absorption of concentrated sodium chloride may cause sudden hypernatremia, cardiovascular shock, CNS disorders, extensive hemolysis, cortical necrosis of the kidneys, and severe local tissue necrosis with extravasation. Use extreme caution; see Dilution. ■ Administration of IV solutions can cause fluid and/or solute overload, resulting in dilution of serum electrolyte concentrations, overhydration, congested states, or pulmonary edema. The risk of dilutional states is inversely proportional to the electrolyte concentration (increased fluids may dilute electrolyte concentration). The risk of solute overload causing congested states with peripheral and pulmonary edema is directly proportional to the electrolyte concentration (higher electrolytes pull in fluid, leading to fluid overload). ■ Overly rapid correction of severe hyponatremia (plasma sodium less than 110 to 115 mEq/L) may lead to a neurologic disorder (osmotic demyelination syndrome [central pontine myelinolysis]), which may be irreversible; see Usual Dose.
Monitor:
Maintain accurate intake and output; monitor electrolytes and acid-base balance, especially in prolonged therapy. ■ Monitor vital signs as indicated. ■ Monitor for signs of hyponatremia (sodium less than 135 mEq/L [e.g., disorientation, headache, lethargy, nausea, weakness]). May progress to coma and seizures. ■ Excessive administration of potassium-free solutions may cause hypokalemia. ■ Before and during use of hypertonic or concentrated sodium chloride, determine osmolar concentrations and chloride and bicarbonate content of the serum. Observe patient continuously to prevent pulmonary edema. ■ Hypertonic solutions can cause vein damage; use a small needle and a large vein to reduce venous irritation and avoid extravasation; see Precautions.
Maternal/child:
Category C: safety for use during pregnancy not established; use only if clearly needed. ■ Use caution in breast-feeding. ■ Benzyl alcohol preservative in bacteriostatic sodium chloride has caused toxicity in newborns. Do not use. ■ Is used in pediatric patients. Safety and effectiveness based on similarity of clinical conditions of pediatric and adult populations. Use caution in neonates or very small infants; the volume of fluid may affect fluid and electrolyte balance.
Elderly:
Lower-end initial doses may be indicated in the elderly; see Dose Adjustments. ■ Incidence of adverse reactions may be increased; monitor carefully, especially with renal or cardiac insufficiency with or without CHF. ■ See Precautions.
Drug/lab interactions
High sodium intake may reduce serum lithium concentrations.
Side effects
Fever, hypovolemia, and injection site reactions may occur.
Osmotic demyelination syndrome (central pontine myelinolysis) secondary to too-rapid correction with hypertonic solutions.
Due to sodium excess:
Aggravation of existing acidosis, anorexia, cellular dehydration, deep respiration, disorientation, distension, edema, hydrogen loss, hyperchloremic acidosis, hypertension, increased BUN, nausea, oliguria, potassium loss, pulmonary edema, water retention, weakness. Excessive excretion of crystalloids to maintain normal osmotic pressure will increase excretion of potassium and bicarbonate and further increase acidosis. Other salts (e.g., iodide and bromide) used for therapy will also be excreted rapidly.
Antidote
Discontinue or decrease rate of infusion; notify the physician of side effects. Sodium excess can be treated by sodium restriction and/or use of diuretics or hemodialysis to remove excessive amounts. Observe patient carefully and treat symptomatically. Save balance of fluid for examination.
Sodium ferric gluconate complex
(SO-dee-um FAIR-ick GLUE-koh-nayt KOM-pleks)
Ferrlecit
Antianemic Iron supplement
pH 7.7 to 9.7
Usual dose
Dose is represented in terms of mg of elemental iron. Given as an infusion and may be administered during the dialysis session.
Treatment of iron deficiency in hemodialysis patients:
125 mg (10 mL)/dose. Doses above 125 mg may be associated with a higher incidence and/or severity of adverse events; see Side Effects. A minimum cumulative dose of 1 Gm of elemental iron is required by most patients. May be administered over eight sessions at sequential dialysis treatments to achieve a favorable hemoglobin or hematocrit response. Additional doses as necessary are indicated to maintain target levels of hemoglobin, hematocrit, and laboratory parameters of iron storage within acceptable limits. Use the lowest dose that achieves this goal.
Pediatric dose
1.5 mg/kg (0.12 mL/kg) of elemental iron as a 1-hour infusion. Do not exceed 125-mg dose. Administer at 8 sequential dialysis sessions. See Maternal/Child.
Dose adjustments
Begin at the low end of the dosing range in elderly patients. Consider decreased cardiac, hepatic, or renal function, and concomitant disease or other drug therapy.
Dilution
Available in ampules or vials containing 62.5 mg/5 mL (12.5 mg/mL) of elemental iron.
Therapeutic dose:
125-mg (10-mL) dose may be diluted in 100 mL NS or may be given undiluted; see Rate of Administration.
Pediatric therapeutic dose:
Dilute calculated dose in 25 mL of NS.
Filters:
Specific studies not available from manufacturer. Filter needle not required by FDA for drug approval; however, use of a filter needle to withdraw it from an ampule should not have an adverse effect.
Storage:
Store ampules at CRT. Do not freeze. Use immediately after dilution in NS.
Compatibility
Manufacturer states, “Do not mix with other medications or add to parenteral nutrition solutions.” Known to be compatible only with NS.
Rate of administration
Too-rapid administration may cause hypotension associated with fatigue; light-headedness; malaise; severe pain in the chest, back, flanks, or groin; and weakness.
Therapeutic dose:
Injection (adults only):
A single dose (undiluted) as a slow IV injection. May be given at a rate up to 12.5 mg/min (1 mL/min).
Infusion (adults and pediatric patients):
A single dose as an infusion properly diluted and equally distributed over 60 minutes.
Actions
A stable macromolecular complex in sucrose injection. Contains elemental iron as the sodium salt of a ferric ion carbohydrate complex. Used to replete the total body content of iron. Iron is critical for normal hemoglobin synthesis to maintain oxygen transport and necessary for metabolism and synthesis of DNA and various other processes. Half-life is approximately 1 hour. Doses of 1 mg/day are adequate to replenish losses in healthy non-menstruating adults. Iron complex is not dialyzable.
Indications and uses
Treatment of iron-deficiency anemia in adult patients and pediatric patients 6 years of age or older with chronic kidney disease who are receiving hemodialysis and supplemental erythropoietin therapy (e.g., EPO, Epogen, Procrit); see Maternal/Child.
Contraindications
All anemias not associated with iron deficiency. Hypersensitivity to sodium ferric gluconate complex or any of its components.
Precautions
Use only when truly indicated to avoid excess storage of iron. Not recommended for use in patients with iron overload. ■ Too-rapid administration may cause clinically significant hypotension; see Rate of Administration and Side Effects. ■ Studies have included patients who have had prior iron dextran exposure with hypersensitivity reactions to at least one form of iron dextran (InFeD or DexFerrum). The majority of these patients tolerated Ferrlecit therapy without a subsequent hypersensitivity reaction. ■ There have been rare occurrences of severe hypersensitivity reactions, including anaphylactic-type reactions, some of which have been life threatening and fatal. Administer in a facility equipped to monitor the patient and respond to any medical emergency. ■ Serum iron levels greater than 300 mcg/dL combined with transferrin oversaturation may indicate iron poisoning; see Side Effects.
Monitor:
Recumbent position during and after injection may help to prevent postural hypotension. Hypotensive effects may be additive to transient hypotension during dialysis and/or from too-rapid rate of infusion; monitor closely. Hypotensive reactions associated with fatigue; light-headedness; malaise; severe pain in the back, chest, flanks, or groin; and weakness may occur with IV iron. These symptoms may or may not be indicative of a hypersensitivity reaction and usually resolve within 1 or 2 hours. ■ Observe continuously for a hypersensitivity reaction during an infusion and after administration for at least 30 minutes and until clinically stable. Monitor vital signs. ■ Periodic monitoring of hemoglobin and hematocrit and iron storage levels recommended. Doses in excess of iron needs may lead to accumulation of iron in iron storage sites and iatrogenic hemosiderosis.
Patient education:
Report S/S of a hypersensitivity reaction (e.g., difficulty breathing, rash, shortness of breath) promptly. Report pain at injection site.
Maternal/child:
Category B: use during pregnancy only if potential benefit justifies potential risk to fetus. ■ Safety for use during breast-feeding and in pediatric patients under 6 years of age not established. ■ Contains benzyl alcohol; should not be used in neonates. Benzyl alcohol present in maternal serum may cross into human milk and may be orally absorbed by a breast-feeding infant.
Elderly:
No age differences identified. Caution in the elderly suggested; see Dose Adjustments.
Drug/lab interactions
May reduce absorption of concomitantly administered oral iron preparations. ■ Concurrent administration of iron therapy with dimercaprol (BAL in Oil) will result in the formation of a toxic complex. Either postpone iron therapy to at least 24 hours after dimercaprol or consider transfusions.
Side effects
Hypotension associated with fatigue; light-headedness; malaise; severe pain in the chest, back, flanks, or groin; and weakness may be caused by too-rapid infusion. Severe hypersensitivity reactions (e.g., angioedema, bronchospasm, cardiac arrest, cardiovascular collapse, dyspnea, edema [oral or pharyngeal], muscle spasm, pain [back or chest], pruritus, urticaria) have been reported rarely. Most commonly reported side effects include abdominal pain, back pain, chest pain, cramps, diarrhea, dizziness, dyspnea, fever, headache, hypersensitivity reactions, hypertension, hypotension, infection, injection site reactions, nausea and vomiting, pain, pharyngitis, pruritus, rhinitis, tachycardia, and thrombosis. Many other side effects occurred in less than 1% of patients and may or may not be attributable to sodium ferric gluconate complex.
Post-marketing:
Anaphylaxis, convulsions, dysgeusia, hypoesthesia, loss of consciousness, pallor, phlebitis, shock, and skin discoloration have been identified. In addition, post-marketing reports have identified that individual doses exceeding 125 mg may result in a higher incidence and/or severity of side effects, including abdominal pain, chest pain, diarrhea, dizziness, dyspnea, hypotension, nausea, paresthesia, peripheral swelling, and urticaria.
Overdose:
Serum iron levels greater than 300 mcg/dL may indicate iron poisoning. May result in hemosiderosis, and excess iron may increase susceptibility to infection. If acute toxicity is seen, it may present as abdominal pain, diarrhea, or vomiting progressing to pallor or cyanosis, lassitude, drowsiness, hyperventilation due to acidosis, iatrogenic hemosiderosis, and cardiovascular collapse.
Antidote
Reduce rate or temporarily discontinue infusion for hypotension; volume expanders (e.g., albumin, hetastarch [Hespan]) may be indicated. Restart when resolved. Discontinue drug and treat hypersensitivity reactions or resuscitate as necessary; notify physician. Epinephrine (Adrenalin) and diphenhydramine (Benadryl) should always be available. In overdose, monitor CBC, iron studies, vital signs, blood gases, and glucose and electrolytes. Maintain fluid and electrolyte balance. Correct acidosis with sodium bicarbonate. Iron complex is not dialyzable.
Sodium phenylacetate and sodium benzoate
(SO-dee-um fen-ill-AH-seh-tate and SO-dee-um BEN-zoh-ate)
Ammonul
Ammonia detoxicant
pH 6 to 8
Usual dose
Must be diluted and administered through a central line. Administration through a peripheral line may cause burns. Given in combination with arginine (dose is dependent on the specific urea cycle disorder [UCD] and is contraindicated in patients with arginase deficiency); see Dilution, Rate of Administration, Precautions, and Monitor. Urea cycle disorders can result from decreased activity of any of the following enzymes: N-acetylglutamate synthetase (NAGS), carbamyl phosphate synthetase (CPS), argininosuccinate synthetase (ASS), ornithine transcarbamylase (OTC), argininosuccinate lyase (ASL), or arginase (ARG); dose and treatment may vary for each. Sodium phenylacetate and sodium benzoate (AMMONUL) infusion should be started as soon as the diagnosis of hyperammonemia is made.
Discontinue analogous oral drugs (e.g., sodium phenylbutyrate [Buphenyl]) before sodium phenylacetate and sodium benzoate (AMMONUL) infusion.
A loading dose as an infusion is administered over 90 to 120 minutes, followed by an equivalent maintenance dose as an infusion administered over 24 hours. Treatment also requires caloric supplementation and restriction of dietary protein. Nonprotein calories should be supplied principally as glucose (8 to 10 mg/kg/min), with intravenous fat (e.g., Intralipid) added to maintain a caloric intake of greater than 80 cal/kg/day.
The dose of sodium phenylacetate and sodium benzoate (AMMONUL) is based on mg/kg in neonates, infants, and younger pediatric patients weighing up to 20 kg and on body surface area for older pediatric patients, adolescents, and adults weighing more than 20 kg as described in the following chart.
| Dose and Administration Guidelines for Sodium Phenylacetate and Sodium Benzoate (AMMONUL) | |||||
| Patient Population | Components of Infusion Solution AMMONUL must be diluted with D10W at equal to or greater than 25 mL/kg before administration | Dosage Provided | |||
| Ammonul | Arginine HCl Injection, 10% | Sodium Phenylacetate | Sodium Benzoate | Arginine HCl | |
| Weight Equal to or Less Than 20 kg: | |||||
| CPS and OTC Deficiency | |||||
| Loading dose: Over 90 to 120 minutes Maintenance: Over 24 hours | 2.5 mL/kg | 2 mL/kg | 250 mg/kg | 250 mg/kg | 200 mg/kg |
| ASS and ASL Deficiency | |||||
| Loading dose: Over 90 to 120 minutes Maintenance: Over 24 hours | 2.5 mL/kg | 6 mL/kg | 250 mg/kg | 250 mg/kg | 600 mg/kg |
| Weight More Than 20 kg: | |||||
| CPS and OTC Deficiency | |||||
| Loading dose: Over 90 to 120 minutes Maintenance: Over 24 hours | 55 mL/M2 | 2 mL/kg | 5.5 Gm/M2 | 5.5 Gm/M2 | 200 mg/kg |
| ASS and ASL Deficiency | |||||
| Loading dose: Over 90 to 120 minutes Maintenance: Over 24 hours | 55 mL/M2 | 6 mL/kg | 5.5 Gm/M2 | 5.5 Gm/M2 | 600 mg/kg |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


