Cyramza
Recombinant monoclonal antibody
Angiogenesis inhibitor
Antineoplastic
pH 6
Usual dose
Premedication:
Before each infusion, premedicate all patients with an intravenous histamine H1 antagonist (e.g., diphenhydramine [Benadryl]). For patients who have experienced a Grade 1 or 2 infusion reaction, also premedicate with dexamethasone (or equivalent) and acetaminophen before each infusion.
Ramucirumab dose:
Gastric cancer:
8 mg/kg every 2 weeks administered as an infusion over 60 minutes. Continue until disease progression or unacceptable toxicity. May be given as a single agent or in combination with paclitaxel. Administer prior to paclitaxel infusion. (In studies, paclitaxel 80 mg/M2 was administered on Days 1, 8, and 15 of each 28-day cycle.)
NSCLC:
10 mg/kg administered as an infusion over 60 minutes on Day 1 of a 21-day cycle. Administer prior to docetaxel infusion. (Docetaxel dose was 60 to 75 mg/M2 in studies.) Continue until disease progression or unacceptable toxicity.
Colorectal cancer:
8 mg/kg administered every 2 weeks as an infusion over 60 minutes before FOLFIRI (irinotecan, folinic acid, and 5-fluorouracil) administration. Continue until disease progression or unacceptable toxicity.
Dose adjustments
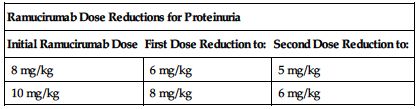
No dose adjustments are recommended for patients with impaired renal function or mild to moderate impaired hepatic function; see Precautions. ■ Permanently discontinue for Grade 3 or 4 infusion-related reactions (IRR), urine protein levels greater than 3 Gm/24 hr, or in the setting of nephrotic syndrome, severe hypertension that cannot be controlled with antihypertensive therapy, GI perforation, Grade 3 or 4 bleeding, or an arterial thromboembolic event. ■ Interrupt therapy for severe hypertension until controlled with medical management. ■ Interrupt ramucirumab therapy before scheduled surgery until the wound is fully healed. ■ Interrupt therapy for urine protein levels 2 Gm/24 hr or higher. Once urine protein level returns to less than 2 Gm/24 hr, reinitiate treatment at a reduced dose as outlined in the following chart. If a urine protein level of 2 Gm/24 hr or higher recurs, interrupt therapy and further reduce the dose as outlined in the following chart once the urine protein level returns to less than 2 Gm/24 hr.
| Ramucirumab Dose Reductions for Proteinuria | ||
| Initial Ramucirumab Dose | First Dose Reduction to: | Second Dose Reduction to: |
| 8 mg/kg | 6 mg/kg | 5 mg/kg |
| 10 mg/kg | 8 mg/kg | 6 mg/kg |
■ For toxicities related to paclitaxel, docetaxel, or the components of FOLFIRI, refer to current prescribing information. ■ See Rate of Administration, Precautions, Monitor, and Antidote.
Dilution
Available as a 10 mg/mL solution in 10- or 50-mL single-use vials. Calculate desired dose and choose the appropriate vial or combination of vials. Withdraw the required volume of ramucirumab and further dilute only with NS to a final volume of 250 mL. Gently invert container to ensure adequate mixing. Do not shake infusion solution.
Filters:
Administration through a protein-sparing, 0.22-micron filter is recommended.
Storage:
Store in original carton in refrigerator at 2° to 8° C (36° to 46° F). Protect from light. Do not shake or freeze. Diluted solutions may be stored for up to 24 hours refrigerated or for up to 4 hours at RT. Contains no preservatives; unused portions must be discarded.
Compatibility
Manufacturer states, “Do not use dextrose-containing solutions. Do not dilute with other solutions or co-infuse with other electrolytes or medications.” Administer through a separate infusion line and flush the line with NS at the end of the infusion.
Rate of administration
Do not administer as an IV push or bolus. Use of a separate line and administration through a protein-sparing, 0.22-micron filter recommended. Administer as an infusion via an infusion pump evenly distributed over 60 minutes. Reduce rate of infusion by 50% for Grade 1 or 2 infusion-related reactions. Flush line with NS at end of infusion.
Actions
A recombinant human IgG1 monoclonal antibody that specifically binds to vascular endothelial growth factor receptor 2 (VEGFR2). Acts as an antagonist, blocking the binding of VEGFR ligands, VEGF-A, VEGF-C, and VEGF-D. As a result, it inhibits ligand-stimulated activation of VEGF Receptor 2, thereby inhibiting ligand-induced proliferation and migration of human endothelial cells. Ramucirumab inhibits angiogenesis in an in vivo animal model. Mean elimination half-life was 15 to 23 days.
Indications and uses
As a single agent or in combination with paclitaxel for the treatment of patients with advanced or metastatic, gastric or gastroesophageal junction adenocarcinoma with disease progression during or after prior fluoropyrimidine- or platinum-containing chemotherapy. ■ In combination with docetaxel for the treatment of patients with metastatic non–small-cell lung cancer (NSCLC) with progression during or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression during FDA-approved therapy for these aberrations before receiving ramucirumab. ■ In combination with FOLFIRI (irinotecan, folinic acid, and 5-fluorouracil) for treatment of patients with metastatic colorectal cancer (mCRC) with progression on or after therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine.
Contraindications
Manufacturer states, “No known contraindications.” However, ramucirumab must be discontinued if an arterial thrombotic event, Grade 3 or 4 bleeding, a Grade 3 or 4 infusion-related reaction, GI perforation, greater than 3 Gm urinary protein/24 hr, nephrotic syndrome, hypertensive crisis, or reversible posterior leukoencephalopathy syndrome (RPLS) develops.
Precautions
Do not administer as an IV push or bolus. ■ Should be administered by or under the direction of a physician specialist in a facility equipped to monitor the patient and respond to any medical emergency. ■ Ramucirumab increases the risk of hemorrhage and gastrointestinal hemorrhage, including severe and sometimes fatal hemorrhagic events. Patients with gastric cancer receiving NSAIDs were excluded from clinical trials; therefore the risk of gastric hemorrhage in these patients is unknown. Patients with NSCLC receiving therapeutic anticoagulation, chronic therapy with NSAIDs, or other antiplatelet therapy (other than a once-daily aspirin) and patients with radiographic evidence of major airway or blood vessel invasion or intra-tumor cavitation were excluded from studies; therefore the risk of pulmonary hemorrhage in these patients is unknown. ■ Serious and sometimes fatal arterial thromboembolic events, including MI, cardiac arrest, cerebrovascular accident, and cerebral ischemia, have been reported. ■ An increased incidence of severe hypertension occurred in patients receiving ramucirumab. Control hypertension before initiating therapy; see Monitor and Dose Adjustment. ■ Infusion reactions have occurred and can be severe. The majority of reactions occurred during or after the first or second infusion. Premedication is recommended; see Usual Dose, Monitor, and Antidote. ■ Ramucirumab is an antiangiogenic therapy that can increase the risk of GI perforation, a potentially fatal event. GI perforation was reported during clinical trials. Consider GI perforation in any patient with complaints of abdominal pain associated with constipation, fever, nausea, and vomiting. ■ Ramucirumab has not been studied in patients with serious or nonhealing wounds. However, as an antiangiogenic therapy it has the potential to adversely affect wound healing. Impaired wound healing can occur with antibodies inhibiting the VEGF pathway. Withhold ramucirumab therapy before surgery. Resume following the surgical intervention based on clinical judgment of adequate wound healing. If a patient develops wound-healing complications during therapy, discontinue ramucirumab until the wound is fully healed. ■ Use with caution and only if potential benefit outweighs risk in patients with Child-Pugh Class B or C cirrhosis. Clinical deterioration manifested by new-onset or worsening encephalopathy, ascites, or hepatorenal syndrome has been reported in these patients. ■ Reversible posterior leukoencephalopathy syndrome (RPLS) has been reported. May present with blindness and other visual and neurologic disturbances, confusion, headache, lethargy, and seizures. Mild to severe hypertension may be present. MRI is required to confirm diagnosis. Symptoms usually resolve gradually with discontinuation of ramucirumab and treatment of hypertension; however, some patients with RPLS experienced ongoing neurologic sequelae or death. ■ Proteinuria occurred during studies; however, it occurred more frequently in patients treated with ramucirumab in combination with FOLFIRI; see Dose Adjustments. ■ A protein substance, it has the potential to produce an immune response. Anti-ramucirumab antibodies have been detected in a small number of patients; clinical significance is not known. ■ See Antidote.
Monitor:
Obtain baseline BP, CBC with differential and platelets, electrolytes, liver function tests, thyroid function tests, and urinalysis. ■ Monitor for S/S of an infusion reaction (e.g., back pain/spasms, chest pain or tightness, chills, dyspnea, flushing, hypoxia, paresthesia, rigors/tremors, wheezing). In severe cases, S/S have included bronchospasm, hypotension, and supraventricular tachycardia. ■ Monitor VS and BP at least every 2 weeks; monitor more frequently in patients with hypertension. ■ Repeat CBC with differential and platelets and electrolytes as indicated. ■ Monitor proteinuria by urine dipstick and/or urinary protein creatinine ratio for the development of worsening proteinuria; see Dose Adjustments. ■ Check surgical wounds for wound dehiscence and monitor for S/S of bleeding or GI perforation (e.g., abdominal pain, constipation, fever, hypotension, nausea and vomiting). ■ Monitor thyroid function during treatment; hypothyroidism has been reported. ■ Monitor for thrombocytopenia (platelet count less than 50,000 mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ See Dose Adjustments, Rate of Administration, Precautions, and Antidote.
Patient education:
Avoid pregnancy during treatment and for at least 3 months after the last dose of ramucirumab. Nonhormonal birth control recommended; see Maternal/Child. Women should report a suspected pregnancy immediately. ■ Increases risk of ovarian failure and may impair fertility. ■ Full disclosure of health history is imperative. ■ Do not undergo any type of surgery without first discussing the risk of impaired wound healing with the health care provider. ■ Report any unusual or unexpected symptoms or side effects promptly (e.g., abdominal pain, bleeding from any source, constipation, dyspnea, persistent cough, sudden onset of worsening neurologic function, vomiting, wound separation). ■ Routine monitoring of BP required. ■ See Appendix D, p. 1333.
Maternal/child:
Can cause fetal harm; avoid pregnancy. Animal models link angiogenesis, VEGF, and VEGF receptor 2 (VEGFR2) to critical aspects of female reproduction, embryo-fetal development, and postnatal development. Use effective contraception during treatment with ramucirumab and for at least 3 months after the last dose. ■ May impair fertility. ■ Discontinue breast-feeding during treatment with ramucirumab. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
No overall differences in safety or efficacy were observed between elderly patients and younger patients being treated for gastric cancer or mCRC. Hazard ratio for overall survival was slightly increased in elderly treated for NSCLC.
Drug/lab interactions
Drug interaction studies have not been completed. No pharmacokinetic interactions were observed between ramucirumab and paclitaxel or between ramucirumab and docetaxel.
Side effects
Single agent:
The most common side effects observed were diarrhea and hypertension. The most common serious side effects observed were anemia and intestinal obstruction. Less frequently reported side effects included arterial thromboembolic events, clinical deterioration in patients with Child-Pugh Class B or C cirrhosis (ascites, encephalopathy, hepatorenal syndrome), epistaxis, GI perforation, headache, hemorrhage, hyponatremia, impaired wound healing, infusion-related reactions, neutropenia, proteinuria, rash, reversible posterior leukoencephalopathy syndrome (RPLS).
In combination with paclitaxel:
The most common side effects observed were diarrhea, epistaxis, fatigue, and neutropenia. The most common serious side effects were neutropenia and febrile neutropenia. Neutropenia and thrombocytopenia were the most common reasons for discontinuation of therapy. Less frequently reported side effects included GI hemorrhagic events, hypertension, hypoalbuminemia, peripheral edema, proteinuria, sepsis, stomatitis, and thrombocytopenia.
In combination with docetaxel:
The most common side effects observed were asthenia/fatigue, neutropenia, and stomatitis/mucosal inflammation. The most common serious side effects were neutropenia, febrile neutropenia, and pneumonia. Infusion-related reactions and epistaxis were the most common reasons for discontinuation of therapy. Less frequently reported side effects included hypertension, hyponatremia, increased lacrimation, peripheral edema, and thrombocytopenia.
In combination with FOLFIRI:
The most common side effects observed were decreased appetite, diarrhea, epistaxis, neutropenia, and stomatitis. The most common serious side effects were diarrhea, febrile neutropenia, and intestinal obstruction. Neutropenia and thrombocytopenia were the most common reasons for discontinuation of any component of treatment, and gastrointestinal perforation and proteinuria were the most common reasons for permanent discontinuation. Other reported side effects included GI hemorrhage events, hypertension, hypoalbuminemia, hypothyroidism, palmar-plantar erythrodysesthesia syndrome, and peripheral edema.
Antidote
Keep physician informed of all side effects. May constitute a medical emergency or will be treated symptomatically as indicated. Permanently discontinue ramucirumab in patients who experience severe bleeding, in patients with impaired wound healing, and in any of the following: Grade 3 or 4 infusion-related reactions, severe hypertension that cannot be controlled with antihypertensive therapy, or in patients with hypertensive crisis or hypertensive encephalopathy, GI perforation, urine protein level greater than 3 Gm/24 hr or in the setting of nephrotic syndrome, a severe arterial thromboembolic event, or reversible posterior leukoencephalopathy syndrome. Treat these side effects aggressively; see Precautions and Monitor. Treat severe infusion reactions as indicated (e.g., epinephrine [Adrenalin], diphenhydramine [Benadryl], IV fluids, oxygen). Temporarily discontinue if moderate to severe proteinuria (equal to or greater than 2 Gm/24 hr) occurs. Resume therapy when proteinuria is less than 2 Gm/24 hr; see Dose Adjustment. Temporarily discontinue for severe hypertension until controlled with medical management; see Monitor and Precautions.
Ranitidine
(rah-NIH-tih-deen)
Zantac
H2 antagonist
Antiulcer agent
Gastric acid inhibitor
pH 6.7 to 7.3
Usual dose
IV injection or intermittent infusion:
50 mg (2 mL) every 6 to 8 hours. Increase frequency of dose, not amount, if necessary for pain relief. 50 mg every 8 to 12 hours may be used short term to replace an oral dose of 150 mg every 12 hours in patients unable to take oral meds. Do not exceed 400 mg/day.
Continuous infusion:
150 mg may be given as a continuous infusion equally distributed over 24 hours. To maintain intergastric acid secretion rates at 10 mEq/hr or less, dose range may be higher in patients with pathologic hypersecretory syndrome (Zollinger-Ellison). Literature suggests an initial dose of 1 mg/kg/hr. Measure gastric acid output in 4 hours. If above 10 mEq/hr or symptoms recur, adjust dose upward in 0.5 mg/kg/hr increments. Up to 2.5 mg/kg/hr has been used.
Additive for total parenteral nutrition (TPN [unlabeled]):
70% to 100% of an average 24-hour dose has been used equally distributed over 24 hours as a continuous infusion. May be supplemented with intermittent doses as needed.
Perioperatively to prevent pulmonary aspiration during anesthesia (unlabeled):
45 to 50 mg 60 minutes before anesthesia.
Prophylaxis and/or control of GI hemorrhage associated with stress ulcers (unlabeled):
150 mg over 24 hours.
Pediatric dose
Safety for selective use in pediatric patients from 1 month to 16 years of age established; see Indications.
One source suggests:
Infants and other pediatric patients:
2 to 4 mg/kg/24 hr in equally divided doses every 6 to 8 hours (0.5 to 1 mg/kg every 6 hours or 0.67 to 1.3 mg/kg every 8 hours). Do not exceed 50 mg/dose.
Neonatal dose
Safety for use of IV ranitidine in neonates under 1 month of age not established; see Maternal/Child.
2 mg/kg every 12 to 24 hours or as a continuous infusion.
Another source suggests 2 mg/kg/24 hr in equally divided doses every 6 to 8 hours (0.5 mg/kg every 6 hours or 0.67 mg/kg every 8 hours).
Dose adjustments
Dose selection should be cautious in the elderly. ■ If the CrCl is less than 50 mL/min, reduce dose to 50 mg every 18 to 24 hours. Gradually increase to 50 mg every 12 hours, or 6 hours with caution if indicated. Adjust schedule to be given after dialysis.
Dilution
IV injection:
Each vial containing 50 mg (2 mL) must be diluted with 18 mL of NS or other compatible infusion solution for injection (D5W, D10W, LR, 5% sodium bicarbonate). Concentration of solution must be no greater than 2.5 mg/mL. Additional diluent may be used.
Intermittent infusion:
Available premixed as a 1 mg/mL solution in 50 mL. May be given without further dilution. Or each 50 mg may be diluted in 100 mL (0.5 mg/mL) of D5W or other compatible infusion solution and given piggyback. Concentration of solution should be no greater than 0.5 mg/mL. Manufacturer recommends discontinuing primary IV during intermittent infusion to avoid incompatibilities. Do not use premixed plastic containers in series connections; may cause air embolism.
Continuous infusion:
Total daily dose may be diluted in 250 mL of D5W or other compatible infusion solution. For Zollinger-Ellison patients, concentration of solution must be no greater than 2.5 mg/mL. In all situations, avoid any contact with aluminum (e.g., needles) during administration. Inspect for color and clarity. Slight darkening of solution does not affect potency. Compatible in selected TPN solutions for 24 hours (consult pharmacist).
Storage:
Store at CRT protected from light. Stable at room temperature for 48 hours after dilution.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Additives should not be introduced into the premixed solution, and the primary IV should be discontinued during ranitidine infusion.”
Solution:
D51/2NS, IV fat emulsion 10%, selected TNA and TPN solutions; see Dilution.
One source suggests the following compatibilities:
Additive:
See previous comments under Compatibility. Acetazolamide (Diamox), amikacin, aminophylline, ampicillin, chloramphenicol (Chloromycetin), chlorothiazide (Diuril), ciprofloxacin (Cipro IV), clindamycin (Cleocin), colistimethate (Coly-Mycin M), dexamethasone (Decadron), digoxin (Lanoxin), dobutamine, dopamine, doxycycline, epinephrine (Adrenalin), erythromycin (Erythrocin), flumazenil (Romazicon), furosemide (Lasix), gentamicin, heparin, isoproterenol (Isuprel), lidocaine, lincomycin (Lincocin), meropenem (Merrem IV), methylprednisolone (Solu-Medrol), midazolam (Versed), nitroprusside sodium, norepinephrine (Levophed), penicillin G potassium and sodium, potassium chloride (KCl), protamine sulfate, quinidine gluconate, tobramycin, vancomycin, zidovudine (AZT, Retrovir).
Y-site:
Acetaminophen (Ofirmev), acyclovir (Zovirax), aldesleukin (Proleukin), allopurinol (Aloprim), amifostine (Ethyol), aminophylline, anidulafungin (Eraxis), atracurium (Tracrium), aztreonam (Azactam), bivalirudin (Angiomax), cefazolin (Ancef), cefoxitin (Mefoxin), ceftaroline (Teflaro), ceftazidime (Fortaz), ciprofloxacin (Cipro IV), cisatracurium (Nimbex), cladribine (Leustatin), dexmedetomidine (Precedex), diltiazem (Cardizem), dobutamine, docetaxel (Taxotere), dopamine, doripenem (Doribax), doxapram (Dopram), doxorubicin liposomal (Doxil), enalaprilat (Vasotec IV), epinephrine (Adrenalin), esmolol (Brevibloc), etoposide phosphate (Etopophos), fenoldopam (Corlopam), fentanyl, filgrastim (Neupogen), fluconazole (Diflucan), fludarabine (Fludara), foscarnet (Foscavir), furosemide (Lasix), gallium nitrate (Ganite), gemcitabine (Gemzar), granisetron (Kytril), heparin, hetastarch in electrolytes (Hextend), hydromorphone (Dilaudid), idarubicin (Idamycin), labetalol, linezolid (Zyvox), lorazepam (Ativan), melphalan (Alkeran), meperidine (Demerol), midazolam (Versed), milrinone (Primacor), morphine, nicardipine (Cardene IV), nitroglycerin IV, norepinephrine (Levophed), ondansetron (Zofran), oxaliplatin (Eloxatin), paclitaxel (Taxol), pancuronium, pemetrexed (Alimta), piperacillin/tazobactam (Zosyn), procainamide (Pronestyl), propofol (Diprivan), remifentanil (Ultiva), sargramostim (Leukine), tacrolimus (Prograf), telavancin (Vibativ), teniposide (Vumon), theophylline, thiotepa, tigecycline (Tygacil), vecuronium, vinorelbine (Navelbine), warfarin (Coumadin), zidovudine (AZT, Retrovir).
Rate of administration
Too-rapid administration has precipitated rare instances of bradycardia, tachycardia, and PVCs.
IV injection:
Each 50 mg or fraction thereof at a rate not to exceed 4 mL/min diluted solution (20 mL over 5 min).
Intermittent infusion:
Each 50-mg dose over 15 to 20 minutes.
Continuous infusion:
Total daily dose equally distributed over 24 hours. Should not exceed a rate of 6.25 mg/hr (10.7 mL/hr if 150 mg [6 mL ranitidine] is diluted in 250 mL). Use of infusion pump preferred to avoid complications of overdose or too-rapid administration.
Actions
A histamine H2 antagonist, it inhibits both daytime and nocturnal basal gastric acid secretion. It also inhibits gastric acid secretion stimulated by food, histamine, bentazole, and pentagastrin. Not an anticholinergic agent. Does not lower calcium levels in hypercalcemia. Onset of action is prompt and effective for 6 to 8 hours. 5 to 12 times more potent than cimetidine. Half-life is 2 to 2.5 hours. Metabolized in the liver. Excreted in the urine. 70% of a dose is recovered in urine as unchanged drug. Crosses placental barrier. Secreted in breast milk.
Indications and uses
Short-term treatment of intractable duodenal ulcers and pathologic hypersecretory conditions in the hospitalized patient. ■ Treatment of active benign gastric ulcers in those patients unable to take oral medication. ■ Treatment of duodenal ulcers in pediatric patients from 1 month to 16 years of age. Safety for use in pathologic hypersecretory conditions not established. ■ Oral dosing for treatment of duodenal and gastric ulcers, maintenance of healing of duodenal and gastric ulcers, and treatment of GERD and erosive esophagitis has been approved for pediatric patients from 1 month to 16 years of age; see package insert.
Unlabeled uses:
Perioperatively to suppress gastric acid secretion, prevent stress ulcers, and prevent aspiration pneumonitis. ■ Reduce the incidence of GI hemorrhage associated with stress ulcers. ■ Additive to TPN to simplify fluid and electrolyte management (decreases the volume and chloride content of gastric secretions).
Contraindications
Known hypersensitivity to ranitidine or its components.
Precautions
Use antacids concomitantly to relieve pain. ■ Gastric malignancy may be present even though patient’s symptoms improve on ranitidine therapy. ■ Use caution in patients with impaired hepatic or renal function. ■ Avoid use in patients with acute porphyria; may precipitate acute porphyric attacks. ■ Gastric pain and ulceration may recur after medication is stopped. ■ Effects maintained with oral dosage. Total treatment usually discontinued after 6 weeks.
Monitor:
Observe frequently; monitor vital signs and pain levels. ■ Obtain baseline SCr. Monitor periodically during extended course of treatment. ■ Monitor ALT if therapy exceeds 400 mg for over 5 days. ■ Change to oral dose when appropriate. ■ See Drug/Lab Interactions.
Patient education:
Stop smoking or at least avoid smoking after the last dose of the day. ■ May increase blood alcohol levels.
Maternal/child:
Category B: use during pregnancy or breast-feeding only when clearly needed. ■ No significant difference in pharmacokinetic parameter values between pediatric patients over 1 month of age and healthy adults when correction is made for body weight. ■ Safety for use of IV ranitidine in neonates under 1 month of age not established; however, limited data suggest that ranitidine may be useful and safe in increasing gastric pH for infants at risk of GI hemorrhage. ■ Half-life in neonates averages 6.6 hours.
Elderly:
Use caution in dose selection; monitoring of renal function suggested; see Dose Adjustments. ■ Half-life is prolonged (3.1 hours) and total clearance is decreased due to reduced renal function. ■ Differences in response between elderly and younger patients not identified; greater sensitivity of some elderly cannot be ruled out. ■ Agitation, confusion (reversible), depression, and hallucination have been reported.
Drug/lab interactions
Concurrent use with warfarin may result in increased or decreased PT and/or INR. Close monitoring is recommended. ■ High doses of ranitidine may reduce the renal excretion of procainamide; monitoring for procainamide toxicity is suggested if ranitidine doses exceed 300 mg/day. ■ May increase serum concentrations of sulfonylureas (e.g., glipizide); monitor blood glucose and adjust dose as indicated. ■ Increased pH may reduce the antibiotic effectiveness of selected cephalosporins (e.g., cefpodoxime [Vantin], cefuroxime [Zinacef]). ■ Increased pH may impair the absorption of atazanavir (Reyataz) and delavirdine (Rescriptor). ■ Effectiveness of gefitinib (Iressa) may be reduced if coadministered with ranitidine and sodium bicarbonate. ■ Reduces the plasma concentrations and antibiotic effectiveness of enoxacin (Penetrex). ■ Monitor for excessive sedation with concurrent administration of selected benzodiazepines (e.g., midazolam [Versed], triazolam [Halcion]). ■ May potentiate the effects of alcohol. ■ May inhibit gastric absorption of itraconazole (Sporanox) and ketoconazole (Nizoral) and reduce antifungal effects. ■ Clinical effect (inhibition of nocturnal gastric secretions) may be reversed by cigarette smoking. ■ Elevated ALT, slight elevation in SCr, and a false-positive for urine protein with Multistix may occur.
Side effects
Abdominal discomfort, burning and itching at IV site, constipation, diarrhea, headache (severe), and nausea and vomiting are the most common side effects. Hypersensitivity reactions (bronchospasm, fever, rash, eosinophilia) can occur. Acute interstitial nephritis, agitation, alopecia, arthralgias, bradycardia, confusion, depression, dizziness, elevated ALT, erythema multiforme, galactorrhea (rare), gynecomastia (rare), hallucinations, hepatitis (reversible), impotence, insomnia, malaise, muscular pain, pneumonia, PVCs, somnolence, tachycardia, vasculitis, and vertigo occur rarely. See Drug/Lab Interactions.
Antidote
Notify physician of all side effects. May be treated symptomatically or may respond to decrease in frequency of dosage. Discontinue ranitidine if S/S of hepatitis with or without jaundice occur. Resuscitate as necessary for overdose. Hemodialysis or peritoneal dialysis may be indicated in overdose.
Rasburicase 
(ras-BYOUR-ih-kase)
Elitek
Antihyperuricemic
Enzyme, urate-oxidase (recombinant)
Usual dose
Prehydration required; see Monitor.
0.2 mg/kg as a single daily dose each day for up to 5 days. Safety and effectiveness of other schedules have not been evaluated. Dosing beyond 5 days and/or administration of more than one course of rasburicase is not recommended. Alternate unlabeled single- and multiple-dose regimens have been used; see literature.
Pediatric dose
Same as adult dose; see Maternal/Child.
Dose adjustments
No dose adjustments recommended.
Dilution
Available in 1.5- and 7.5-mg vials with manufacturer-supplied diluent (SWFI and Poloxamer 188). Determine the vial size and/or number of vials needed to provide the calculated dose. Reconstitute each 1.5-mg vial with 1 mL of diluent and each 7.5-mg vial with 5 mL of diluent. Final concentration is 1.5 mg/mL. Mix by swirling gently. Do not shake. Withdraw the calculated dose of reconstituted solution and inject into an infusion bag containing the appropriate volume of NS to achieve a final total volume of 50 mL. (For example a 20-kg child would receive a dose of 4 mg [20 kg × 0.2 mg/kg = 4 mg]. Reconstitute 3 vials, each with 1 mL of the diluent provided. Withdraw 2.7 mL of reconstituted solution and add to an infusion bag containing 47.3 mL of NS.)
Filters:
No filters should be used for the infusion.
Storage:
Refrigerate unopened lyophilized drug product and diluent. Do not freeze; protect from light. Both the reconstituted and diluted product may be stored for 24 hours if refrigerated. Discard any unused product.
Compatibility
Manufacturer states, “Should be infused through a different line than that used for the infusion of other concomitant medications. If use of a separate line is not possible, the line should be flushed with at least 15 mL of NS prior to and after infusion with rasburicase.”
Rate of administration
A single dose as an infusion equally distributed over 30 minutes.
Do not administer as a bolus infusion. Do not filter infusion.
Actions
A recombinant urate-oxidase enzyme produced by genetic engineering. Catalyzes enzymatic oxidation of uric acid into an inactive and soluble metabolite (allantoin). Onset of action is 4 hours. Mean terminal half-life is similar between pediatric and adult patients and ranges from 15.7 to 22.5 hours.
Indications and uses
Initial management of plasma uric acid levels in pediatric and adult patients with leukemia, lymphoma, and solid tumor malignancies who are receiving anticancer therapy that is expected to result in tumor lysis and subsequent elevation of plasma uric acid.
Limitation of use:
Indicated only for a single course of treatment.
Contraindications
Glucose-6-phosphatase dehydrogenase (G6PD) deficiency or a history of anaphylaxis or severe hypersensitivity, hemolytic reactions, or methemoglobinemia reactions to rasburicase.
Precautions
May cause severe hypersensitivity reactions, including anaphylaxis. Reactions may occur at any time during treatment, including with the first dose; see Monitor and Antidote. ■ Has caused severe hemolytic reactions in patients with G6PD deficiency. It is recommended that patients at higher risk for G6PD deficiency (e.g., patients of African or Mediterranean descent) be screened before starting rasburicase therapy. See Contraindications and Antidote. ■ Methemoglobinemia has been reported. Patients developed serious hypoxemia, requiring intervention and medical support. See Antidote. ■ As with all therapeutic proteins, there is the potential for immunogenicity. May elicit antibodies that inhibit the activity of rasburicase.
Monitor:
Monitor serum uric acid levels, electrolytes, and renal function before and during therapy. ■ Patients should be hydrated intravenously according to standard medical practice for the management of plasma uric acid in patients at risk for tumor lysis syndrome (TLS). ■ Maintain urine at neutral or slightly alkaline pH. ■ Observe for symptoms of TLS (e.g., hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia). If untreated, may develop acute uric acid nephropathy, leading to renal failure. ■ Monitor for S/S of a hypersensitivity reaction (e.g., anaphylaxis, bronchospasm, chest pain, dyspnea, hypotension, hypoxia, shock, urticaria). ■ Screen patients at risk for hemolysis; see Contraindications and Precautions. Monitor for S/S of hemolysis (e.g., anemia, jaundice with increased indirect bilirubin and LDH, pallor, reduced haptoglobin). In studies, severe hemolytic reactions occurred within 2 to 4 days of the start of therapy. ■ Monitor for S/S of methemoglobinemia (e.g., cyanosis, dyspnea, headache, hypoxemia, lethargy, methemoglobin). ■ Will cause enzymatic degradation of uric acid within blood samples left at room temperature, resulting in falsely low uric acid levels. To ensure accurate measurement, blood must be collected into prechilled tubes containing heparin anticoagulant and immediately immersed and maintained in an ice water bath. Plasma samples must be prepared by centrifugation in a precooled centrifuge (4° C [39° F]). Plasma samples must be analyzed within 4 hours of sample collection.
Patient education:
Report blood in urine, painful urination, or signs of a hypersensitivity reaction promptly.
Maternal/child:
Category C: studies have not been performed. Potential benefits must justify potential risks to fetus. ■ Discontinue breast-feeding. ■ Studied in pediatric patients from 1 month to 17 years of age. Pediatric patients under 2 years of age had a higher mean uric acid AUC and a lower rate of success at achieving normal uric acid concentrations by 48 hours than did older pediatric patients.
Elderly:
No overall differences in pharmacokinetics, safety, and effectiveness were observed between elderly and younger patients.
Drug/lab interactions
Studies have not been conducted. ■ Does not metabolize allopurinol (Aloprim), cytarabine (ARA-C), methylprednisolone (Solu-Medrol), methotrexate, 6-mercaptopurine (Purinethol), thioguanine, etoposide (VePesid), etoposide phosphate (Etopophos), daunorubicin (Cerubidine), cyclophosphamide, or vincristine in vitro. Metabolic-based drug interactions are not anticipated with these agents. ■ Did not affect the activity of P450 isoenzymes in preclinical in vivo studies. Clinically relevant P450-mediated drug-drug interactions are not anticipated. ■ Will cause enzymatic degradation of uric acid within blood samples left at room temperature, resulting in falsely low uric acid levels; see Monitor.
Side effects
The most common adverse reactions are abdominal pain, anxiety, constipation, diarrhea, fever, headache, nausea, peripheral edema, and vomiting. Serious adverse reactions observed are hemolysis, hypersensitivity reactions (e.g., anaphylaxis, chest pain, dyspnea, hypotension, urticaria), methemoglobinemia, and severe rash. Other observed reactions include abdominal and GI infections, acute renal failure, fluid overload, hyperbilirubinemia, hyperphosphatemia, hypophosphatemia, increased ALT, ischemic coronary artery disorders, mucositis, neutropenia with or without fever, pharyngolaryngeal pain, pulmonary hemorrhage, rash, respiratory distress/failure, sepsis, and supraventricular arrhythmias.
Post-marketing:
Convulsions, muscle contractions (involuntary).
Antidote
Notify physician of all side effects. Should be immediately and permanently discontinued in patients who experience severe hypersensitivity reactions, hemolysis, or methemoglobinemia. Do not rechallenge. Treat anaphylaxis with epinephrine, corticosteroids (e.g., dexamethasone [Decadron]), oxygen, and antihistamines (diphenhydramine [Benadryl]). Hemolysis or methemoglobinemia may require transfusion support. Methylene blue may be required for treatment of methemoglobinemia. There is no specific antidote. Resuscitate as indicated.
Raxibacumab
(rack-see-BACK-u-mab)
ABthrax
Monoclonal antibody
Antidote
Usual dose
Premedication in adults:
Premedicate with diphenhydramine 25 to 50 mg within 1 hour before raxibacumab infusion to prevent or attenuate infusion reactions. May be given IV or PO depending on proximity to the start of the raxibacumab infusion.
Adults:
A single dose of 40 mg/kg as an infusion over 2 hours and 15 minutes.
Pediatric dose
Premedication in pediatric patients:
Premedicate with an appropriate dose of diphenhydramine within 1 hour before raxibacumab infusion to prevent or attenuate infusion reactions. May be given IV or PO depending on proximity to the start of the raxibacumab infusion.
Dosage for pediatric patients:
Weight more than 50 kg:
A single dose of 40 mg/kg.
Weight more than 15 kg and up to 50 kg:
A single dose of 60 mg/kg.
Weight 15 kg or less:
A single dose of 80 mg/kg.
Dose adjustments
No dose adjustments indicated based on age, gender, or race.
Dilution
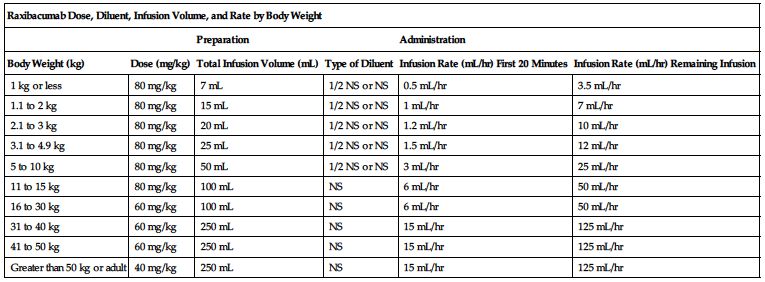
Available as a single-use vial that contains 1700 mg/34 mL (50 mg/mL). The recommended dose is weight-based and is given as an intravenous infusion after appropriate dilution in a compatible solution. The following chart outlines dose, diluent, total infusion volume, and infusion rate based on patient body weight.
| Raxibacumab Dose, Diluent, Infusion Volume, and Rate by Body Weight | |||||
| Preparation | Administration | ||||
| Body Weight (kg) | Dose (mg/kg) | Total Infusion Volume (mL) | Type of Diluent | Infusion Rate (mL/hr) First 20 Minutes | Infusion Rate (mL/hr) Remaining Infusion |
| 1 kg or less | 80 mg/kg | 7 mL | 1/2 NS or NS | 0.5 mL/hr | 3.5 mL/hr |
| 1.1 to 2 kg | 80 mg/kg | 15 mL | 1/2 NS or NS | 1 mL/hr | 7 mL/hr |
| 2.1 to 3 kg | 80 mg/kg | 20 mL | 1/2 NS or NS | 1.2 mL/hr | 10 mL/hr |
| 3.1 to 4.9 kg | 80 mg/kg | 25 mL | 1/2 NS or NS | 1.5 mL/hr | 12 mL/hr |
| 5 to 10 kg | 80 mg/kg | 50 mL | 1/2 NS or NS | 3 mL/hr | 25 mL/hr |
| 11 to 15 kg | 80 mg/kg | 100 mL | NS | 6 mL/hr | 50 mL/hr |
| 16 to 30 kg | 60 mg/kg | 100 mL | NS | 6 mL/hr | 50 mL/hr |
| 31 to 40 kg | 60 mg/kg | 250 mL | NS | 15 mL/hr | 125 mL/hr |
| 41 to 50 kg | 60 mg/kg | 250 mL | NS | 15 mL/hr | 125 mL/hr |
| Greater than 50 kg or adult | 40 mg/kg | 250 mL | NS | 15 mL/hr | 125 mL/hr |
Weight in kg × Dose in mg/kg = Calculated dose of raxibacumab in mg
Calculated dose in mg ÷ Raxibacumab concentration (50 mg/mL) = Required volume of raxibacumab in mL
Based on total infusion volume from the previous chart, prepare either a syringe or infusion bag as appropriate.
Bag preparation:
Select the appropriate size bag of compatible solution. Withdraw a volume of solution from the bag equal to the calculated volume in milliliters of raxibacumab and discard. Withdraw the calculated dose of raxibacumab from the vial and transfer into the prepared infusion bag. Gently invert bag to mix. Do not shake.
Syringe preparation:
Select an appropriate size syringe for the total volume of infusion to be administered. Using this syringe, withdraw the calculated raxibacumab dose. Then withdraw an appropriate amount of compatible solution to prepare a total volume infusion syringe as specified in the chart. Gently mix. Do not shake.
Filters:
Not required by manufacturer; additional data not available.
Storage:
Store in original carton until time of use to protect from light. Refrigerate at 2° to 8° C (36° to 46° F). Do not freeze. Discard unused raxibacumab. Prepared solution stable for 8 hours at RT.
Compatibility
Specific information not available; consider specific use; consult pharmacist.
Rate of administration
Administer as outlined in the chart under Dilution. Total infusion time is 2 hours and 15 minutes. Slow or interrupt the infusion for infusion-related symptoms or other side effects.
Actions
A human IgG1λ monoclonal antibody. Produced by recombinant DNA technology in a murine cell expression system. It does not have direct antibacterial activity but binds to the protective antigen (PA) of B. anthracis toxin. Raxibacumab inhibits the binding of the PA to its cellular receptor. This prevents the intracellular entry of the anthrax lethal factor and edema factor, the enzymatic toxin components responsible for the pathogenic effects of the anthrax toxin. Mean steady-state volume of distribution was greater than plasma volume, suggesting some tissue distribution. Virtually no renal clearance occurs.
Indications and uses
Treatment of adult and pediatric patients with inhalation anthrax due to Bacillus anthracis in combination with the appropriate antibacterial drugs. ■ Prophylaxis of inhalation anthrax when alternative therapies are not appropriate or available for use.
Limitations of use:
Effectiveness based solely on efficacy studies in animal models of inhalational anthrax. ■ Does not cross the blood-brain barrier and does not prevent or treat meningitis. ■ Does not have direct antibacterial activity. Should be used in combination with appropriate antibacterial drugs. ■ See Maternal/Child.
Contraindications
Manufacturer states, “None.”
Precautions
Infusion-related reactions have been reported; see Monitor and Antidote. ■ A protein product; has the potential for immunogenicity. ■ Adequate laboratory and supportive medical resources must be available.
Monitor:
Premedication required; see Usual Dose. ■ Monitor for infusion-related reactions during administration (e.g., dyspnea, pruritus, rash, urticaria) and slow or interrupt infusion as indicated.
Patient education:
Promptly report S/S of an infusion reaction (e.g., hives, itching, rash, shortness of breath). ■ The effectiveness of raxibacumab is based on studies done in animals and has not been tested in humans with anthrax.
Maternal/child:
Category B: safety for use in pregnancy, breast-feeding, and pediatric patients not established. Use raxibacumab only if clearly needed. ■ As in adults, effectiveness in pediatric patients is based solely on efficacy studies in animal models. There are no studies of safety or pharmacokinetics in pediatric patients.
Elderly:
Numbers in clinical studies insufficient to determine if the elderly respond differently compared with younger subjects.
Drug/lab interactions
The pharmacokinetics of ciprofloxacin and raxibacumab was not altered with coadministration of 40 mg/kg raxibacumab IV and either IV or oral ciprofloxacin.
In a study determining the efficacy of the drugs in the treatment of animals with systemic anthrax disease, the pharmacokinetics of levofloxacin and raxibacumab was unaffected by product coadministration.
Side effects
The most frequently reported side effects were pain in extremities, pruritus, rash, and somnolence. Anemia, back pain, dizziness, fatigue, flushing, hypertension, increased blood amylase and blood creatine phosphokinase, infusion site pain, insomnia, leukopenia, lymphadenopathy, muscle spasms, palpitations, peripheral edema, prolonged PT, and vasovagal syncope have been reported.
Antidote
Notify physician of all side effects. Most will be treated symptomatically. If signs of an infusion reaction occur, slow or interrupt the infusion and treat appropriately. No cases of overdose have been observed. If overdose occurs, monitor for any signs or symptoms of adverse effects and apply appropriate medical therapies.
Reslizumab 
Cinqair
Interleukin-5 antagonist
Monoclonal antibody (IgG4κ)
pH 5.5
Usual dose
3 mg/kg once every 4 weeks as an infusion over 20 to 50 minutes.
Dose adjustments
No dose adjustments required based on age, gender, or race. Clinical studies have not been conducted to assess the effect of hepatic or renal impairment on the pharmacokinetics of reslizumab.
Dilution
Supplied as a clear to slightly hazy/opalescent, colorless to slightly yellow solution in single-use vials containing 100 mg/10 mL (10 mg/mL). May contain a few translucent-to white amorphous particulates. Do not shake. Aseptic technique required. Allow solution to reach room temperature. Withdraw the proper volume of reslizumab from the vial(s) based on the recommended weight-based dose and slowly add to a 50-mL infusion bag of NS. Gently invert to mix the solution. Do not shake.
Filters:
Use of an infusion set with an in-line, low–protein-binding, 0.2-micron filter is required. Compatible with polyethersulfone (PES), polyvinylidene fluoride (PVDF), nylon, and cellulose acetate in-line infusion filters.
Storage:
Before use, refrigerate at 2° to 8° C (36° to 46° F) in original carton to protect from light. Do not freeze. Do not shake. Administer diluted solution immediately after preparation or may be refrigerated or kept at RT (up to 25° C [77° F]), protected from light, for up to 16 hours. Time between preparation and administration should not exceed 16 hours. If refrigerated before administration, allow the diluted solution to reach RT. Discard unused portion.
Compatibility
Manufacturer states, “Do not mix or dilute with other drugs. Do not infuse concomitantly in the same IV line with other agents.” Compatible with polyvinylchloride (PVC) or polyolefin infusion bags and with polyethersulfone (PES), polyvinylidene fluoride (PVDF), nylon, and cellulose acetate in-line infusion filters.
Rate of administration
For IV infusion only. Do not administer as an IV push or bolus. Use of an infusion set with an in-line, low–protein-binding, 0.2-micron filter is required.
Administer a single dose as an infusion over 20 to 50 minutes. Infusion time varies based on weight-based dosing. After administration, flush the IV line with NS to ensure that all of the reslizumab has been administered.
Actions
Inflammation is an important component in the pathogenesis of asthma. Multiple cell types and mediators are involved in inflammation. Reslizumab is a humanized interleukin-5 antagonist monoclonal antibody (IgG4, kappa). IL-5 is the major cytokine responsible for the growth and differentiation, recruitment, activation, and survival of eosinophils. Reslizumab binds to IL-5, inhibiting the bioactivity of IL-5. By inhibiting IL-5 signaling, reslizumab reduces the production and survival of eosinophils, one of the cell types implicated in the inflammation seen with asthma. Specific mechanism of action not definitively established. Following administration of reslizumab, reductions in blood eosinophil counts were observed and maintained through 52 weeks of treatment. There is minimal distribution into the extravascular tissues. Reslizumab is degraded by enzymatic proteolysis into small peptides and amino acids. Half-life is approximately 24 days.
Indications and uses
Add-on maintenance treatment of patients with severe asthma who have an eosinophilic phenotype and are 18 years of age or older.
Limitation of use:
Not indicated for the treatment of other eosinophilic conditions. ■ Not indicated for the relief of acute bronchospasm or status asthmaticus.
Contraindications
Known hypersensitivity to reslizumab or any of its excipients.
Precautions
For IV use only. ■ Administered by or under the direction of a physician knowledgeable in its use and in a facility equipped to monitor the patient and respond to any medical emergency. ■ Hypersensitivity reactions, including anaphylaxis, have been reported. ■ Should not be used to treat acute asthma symptoms or acute exacerbations. Do not use to treat acute bronchospasm or status asthmaticus. ■ Malignant neoplasms have been reported. The majority were diagnosed within less than 6 months of exposure to reslizumab. ■ No clinical studies have been conducted to assess the reduction of maintenance corticosteroid doses following administration of reslizumab. Do not discontinue systemic or inhaled corticosteroids abruptly upon initiation of reslizumab therapy. ■ Eosinophils may be involved in the immunologic response to some helminth infections. The effects of reslizumab on the immune response against parasitic infections are unknown. Patients with known parasitic infections were excluded from clinical studies. ■ A therapeutic protein, there is a potential for immunogenicity. ■ See Monitor.
Monitor:
Monitor for S/S of a hypersensitivity reaction during and following completion of the infusion. In clinical trials, anaphylaxis was observed during or within 20 minutes after completion of the infusion and was reported as early as the second dose of reslizumab. Manifestations included decreased oxygen saturation, dyspnea, skin and mucosal involvement (including urticaria), vomiting, and wheezing. ■ Reductions in corticosteroid dose, if appropriate, should be gradual and under physician supervision. Monitor for systemic withdrawal symptoms and/or conditions previously suppressed by systemic corticosteroid therapy. ■ Treat pre-existing helminth infections before initiating reslizumab. Discontinue treatment until infection resolves in patients who become infected during therapy and do not respond to anti-helminth treatment.
Patient education:
Immediately report any S/S of a hypersensitivity reaction (e.g., chest discomfort, cough, dyspnea, postural dizziness, pruritus, rash, throat irritation, urticaria, wheezing) occurring during or after administration. ■ Does not treat acute asthma symptoms or acute exacerbations. Seek medical advice if asthma remains uncontrolled or worsens after initiation of treatment with reslizumab. ■ Malignancies have been reported. ■ Do not discontinue or reduce the dose of maintenance systemic or inhaled corticosteroids except under the direct supervision of a physician.
Maternal/child:
Use during pregnancy only if clearly needed. In women with poorly or moderately controlled asthma, evidence demonstrates an increased risk of pre-eclampsia in the mother and an increased risk of prematurity, low birth weight, and smaller for gestational age in the neonate. The level of asthma control should be closely monitored in pregnant women and treatment adjusted as necessary to maintain optimal control. Monoclonal antibodies such as reslizumab cross the placental barrier. The potential effects on a fetus are likely to be greater during the second and third trimester of pregnancy. Consider the long half-life of reslizumab. ■ Use caution during breast-feeding; effects unknown. ■ Safety and effectiveness for use in pediatric patients 17 years of age or younger not established.
Elderly:
No overall differences in safety or effectiveness observed between younger patients and patients 65 years of age and older. No dose adjustment is necessary.
Drug/lab interactions
No formal drug interaction studies have been performed. Population pharmacokinetics analyses indicate that concomitant use of either leukotriene antagonists (e.g., montelukast [Singulair]) or corticosteroids does not affect the pharmacokinetics of reslizumab.
Side effects
Oropharyngeal pain is the most common side effect. Less commonly reported adverse reactions include myalgia and transient creatine phosphokinase (CPK) elevations. Hypersensitivity reactions (including anaphylaxis) and malignancy are the most serious side effects.
Antidote
Keep the physician informed of all side effects. Most will be treated symptomatically. If a hypersensitivity reaction occurs, discontinue the infusion immediately and treat with oxygen, epinephrine, antihistamines (e.g., IV diphenhydramine [Benadryl]), corticosteroids, albuterol, vasopressors (e.g., dopamine), and ventilation equipment as indicated. Symptoms of overdose were not noted in clinical trials. If overdose occurs, monitor patients for S/S of adverse effects. Resuscitate as necessary.
Reteplase recombinant
(REE-teh-place re-KOM-buh-nant)
Retavase, r-PA
Thrombolytic agent (recombinant)
pH 7 to 7.4
Usual dose
Administered concomitantly with heparin. Give a 5,000-unit IV bolus of heparin before the initial injection of reteplase, then give 10 units (10 mL) of reteplase as an IV injection. Follow with a 1,000 unit/hr continuous IV infusion of heparin for at least 24 hours. Give a second 10-unit bolus of reteplase 30 minutes after the first. See Dilution, Compatibility, and Rate of Administration. Aspirin is also used either during or following heparin treatment; an initial dose of 160 to 350 mg is followed by doses of 75 to 350 mg.
Dose adjustments
The second bolus should not be given if serious bleeding in a critical location (e.g., intracranial, gastrointestinal, retroperitoneal, pericardial) occurs before it is due to be given.
Dilution
Supplied in a kit with all components for reconstitution. Each kit contains a package insert and two of each of the following: single-use reteplase vials (10.8 units each), single-use diluent vials of SWFI (10 mL each), sterile 10-mL syringes with 20-gauge needles attached, sterile dispensing pins, sterile 20-gauge needles for administration, and alcohol swabs. Withdraw diluent with 20-gauge needle. Discard needle and put dispensing pin on syringe of diluent. Transfer diluent to vial of reteplase. Pin and syringe should remain in place while vial is swirled to dissolve reteplase. Do not shake. When completely dissolved, withdraw 10 mL reconstituted solution into the syringe (vials are 0.7 mL overfilled). Remove dispensing pin and replace with a 20-gauge needle for administration.
Storage:
Kit should remain sealed to protect contents from light. Store at 2° to 25° C (36° to 77° F). Do not use beyond expiration date. Contains no preservatives; should be reconstituted immediately before use, but may be stored at room temperature if used within 4 hours. Discard all unused solution and supplies.
Compatibility
Manufacturer states, “Should be given via an IV line in which no other medication is being simultaneously injected or infused. No other medication should be added to the injection solution containing reteplase. Incompatible with heparin; do not administer heparin in the same IV line unless the line is flushed through with NS or D5W before and after reteplase.”
Rate of administration
Heparin:
First 1,000 units over 1 minute. After this test dose, the balance of 4,000 units may be given over 1 minute. Follow with an infusion of 1,000 units/hr.
Reteplase:
A single dose evenly distributed over 2 minutes. To avoid incompatibilities and ensure delivery of both doses, be sure to flush line with a minimum of 30 to 50 mL NS or D5W before and after each injection.
Actions
A recombinant plasminogen activator. Exerts its thrombolytic action by generating plasmin from plasminogen through a specific process. Plasmin then degrades the fibrin matrix of the thrombus. Potency is expressed in units that are specific to reteplase. With therapeutic doses, a decrease in circulating fibrinogen makes the patient susceptible to bleeding. Onset of action is prompt, effecting patency of the vessel within 90 minutes in most patients. The FDA has allowed the manufacturer to claim superiority over alteplase at achieving patency within 90 minutes. Prompt opening of arteries increases probability of improved cardiac function. Half-life is 13 to 16 minutes. Cleared from the plasma by the liver and kidneys. Mean fibrinogen level should return to baseline value within 48 hours.
Indications and uses
Management of acute myocardial infarction (AMI) in adults for the improvement of ventricular function following AMI, the reduction of the incidence of congestive failure, and the reduction of mortality associated with AMI. Treatment should begin as soon as possible after the onset of symptoms of AMI. ■ Current AHA and JAMA recommendations identify thrombolytic agents as Class I therapy in patients younger than 70 years with recent onset of chest pain (within 6 hours) consistent with AMI and at least 0.1 mV of ST segment elevation in at least two ECG leads. Use in all other patients based on age, accurate diagnosis, and time from onset of chest pain.
Contraindications
Active internal bleeding, arteriovenous malformation or aneurysm, bleeding diathesis, history of cerebral vascular accident, intracranial or intraspinal surgery or trauma within 2 months, intracranial neoplasm, severe uncontrolled hypertension.
Precautions
Administered under the direction of a physician knowledgeable in its use and with appropriate emergency drugs and diagnostic and laboratory facilities available. ■ Reperfusion arrhythmias occur frequently (e.g., sinus bradycardia, accelerated idioventricular rhythm, PVCs, ventricular tachycardia); have antiarrhythmic medications available at bedside. ■ A greater alteration of hemostatic status than with heparin. Strict bed rest indicated to reduce risk of bleeding. Use extreme care with the patient; avoid any excessive or rough handling or pressure (including too-frequent BPs); avoid invasive procedures (e.g., arterial puncture, venipuncture, IM injection). If these procedures are absolutely necessary, use extreme precautionary methods (use radial artery instead of femoral; small-gauge catheters and needles, and sites that are easily observed and compressible where bleeding can be controlled; avoid handling of catheter sites, and use extended pressure application of up to 30 minutes). Minor bleeding occurs often at catheter insertion sites. Avoid use of razors and toothbrushes. ■ Use extreme caution and weigh risks against anticipated benefits in the following situations: recent major surgery (e.g., coronary artery bypass graft, obstetric delivery, organ biopsy), previous puncture of noncompressible vessels (e.g., jugular, subclavian), cerebrovascular disease, recent GI or GU bleeding, recent trauma, hypertension (e.g., systolic BP equal to or greater than 180 mm Hg and/or diastolic BP equal to or greater than 110 mm Hg), high likelihood of left heart thrombus (e.g., mitral stenosis with atrial fibrillation), acute pericarditis, subacute bacterial endocarditis, hemostatic defects including those secondary to severe hepatic or renal disease, severe hepatic or renal dysfunction, pregnancy, diabetic hemorrhagic retinopathy or other hemorrhagic ophthalmic conditions, septic thrombophlebitis or occluded AV cannula at a seriously infected site, advanced age, patients currently receiving oral anticoagulants (e.g., warfarin [Coumadin]), any other condition in which bleeding constitutes a significant hazard or would be particularly difficult to manage because of its location. ■ Simultaneous therapy with continuous infusion of heparin is used to reduce the risk of rethrombosis. Markedly increases risk of bleeding. ■ Standard treatment for myocardial infarction continues simultaneously with reteplase therapy except if temporarily contraindicated (e.g., arterial blood gases unless absolutely necessary). ■ No experience with patients receiving repeat courses of reteplase. ■ Cholesterol embolization has been reported and may be fatal.
Monitor:
Best to establish separate IV lines for reteplase and heparin. If not appropriate, be sure to flush the IV line before and after each injection of reteplase. ■ Baseline ECG, CPK, and clotting studies (TT, PTT, CBC, fibrinogen level, platelets) and baseline assessment (patient condition, pain, hematomas, petechiae, or recent wounds) should be completed before administration. Type and cross-match may also be ordered. ■ Monitor ECG continuously, and record strips with greatest ST segment elevation initially and every 15 minutes for at least 4 hours. A 12-lead ECG is indicated when therapy is complete. ■ Maintain strict bed rest; monitor the patient carefully and frequently for anginal pain and signs of bleeding; observe catheter sites at least every 15 minutes and apply pressure dressings to any recently invaded site; watch for hematuria, hematemesis, bloody stool, petechiae, hematoma, flank pain, muscle weakness; do neuro checks every hour. Continue until normal clotting function returns. ■ Watch for extravasation. ■ See Precautions and Drug/Lab Interactions.
Patient education:
Compliance with all measures to minimize bleeding (e.g., strict bed rest) is very important. ■ Avoid use of razors, toothbrushes, and other sharp items. ■ Use caution while moving to avoid excessive bumping. ■ Report all episodes of bleeding and apply local pressure if indicated. Expect oozing from IV sites.
Maternal/child:
Category C: has resulted in hemorrhage leading to spontaneous abortions in rabbits. Safety for use in pregnancy, breast-feeding, and pediatric patients not established.
Elderly:
See Indications and Precautions. ■ May have poorer prognosis following AMI and pre-existing conditions that may increase risk of intracranial bleeding. Select patients carefully to maximize benefits.
Drug/lab interactions
Interaction of reteplase with other cardioactive drugs has not been studied. Risk of bleeding may be increased by any medicine that affects blood clotting, including anticoagulants (e.g., heparin, warfarin [Coumadin]); any medication that may cause hypoprothrombinemia, thrombocytopenia, or GI ulceration or bleeding (e.g., selected antibiotics [e.g., cefotetan], aspirin, NSAIDs [e.g., ibuprofen (Advil, Motrin), naproxen (Aleve, Naprosyn)]); and/or any other medication that inhibits platelet aggregation (e.g., clopidogrel [Plavix], dipyridamole [Persantine], glycoprotein GPIIb/IIIa receptor antagonists [e.g., abciximab (ReoPro), eptifibatide (Integrilin), tirofiban (Aggrastat)], plicamycin [Mithracin], sulfinpyrazone [Anturane], ticlopidine [Ticlid], valproic acid [Depacon]). Concurrent use not recommended with the exception of heparin and aspirin (in AMI) to reduce the risk of rethrombosis. If concurrent or subsequent use is indicated (e.g., management of acute coronary syndrome, percutaneous coronary intervention), monitor PT and aPTT closely. ■ Coagulation tests will be unreliable; specific procedures can be used; notify the lab of reteplase use.
Side effects
Bleeding is most common: internal (GI tract, GU tract, intracranial, respiratory, or retroperitoneal sites), epistaxis, gingival, and superficial or surface bleeding (venous cutdowns, arterial punctures, sites of recent surgical intervention). Reperfusion arrhythmias are common; other serious arrhythmias may occur. A few hypersensitivity reactions, as well as fever, hypotension, nausea, and vomiting, have occurred. Cholesterol embolism has been reported and may be fatal. Clinical S/S may include acute renal failure, gangrenous digits, hypertension, infarctions (e.g., bowel, cerebral, myocardial, or spinal cord), pancreatitis, “purple toe” syndrome, renal artery occlusion.
Antidote
Notify physician of all side effects. Note even the most minute bleeding tendency. Oozing at IV sites is expected. Control minor bleeding by local pressure. For severe bleeding in a critical location, discontinue second dose of reteplase if it has not been given and any heparin therapy immediately. Whole blood, packed RBCs, cryoprecipitate, fresh frozen plasma, platelets, desmopressin, tranexamic acid, and aminocaproic acid may all be indicated. Topical preparations of aminocaproic acid may stop minor bleeding. Consider protamine if heparin has been used. Treat bradycardia with atropine, reperfusion arrhythmias with lidocaine or procainamide; VT or VF may require cardioversion. Treat minor hypersensitivity reactions symptomatically. Discontinue drug and treat anaphylaxis as indicated; resuscitate as necessary. Discontinue therapy if any symptoms of cholesterol embolism occur.
Rho(D) immune globulin intravenous (human) 
(ih-MUNE GLAW-byoo-lin IN-trah-ve-nes)
Rho(D)-IGIV, Rhophylac, WinRho SDF
Immunizing agent (passive)
Platelet count stimulator
pH 6.5 to 7.6
Usual dose (international units [IU])
Pregnancy, predelivery:
Rhophylac and WinRho SDF:
Confirm Rho(D) negative status of patient. May be given IV or IM. 1,500 IU (300 mcg) at 28 weeks’ gestation. If WinRho SDF is administered early in the pregnancy, it should be repeated at 12-week intervals to maintain an adequate level of passively acquired anti-Rh.
Pregnancy, postdelivery:
Confirm Rho(D) negative status of patient. May be given IV or IM. Administer as soon as possible after delivery of a confirmed Rho(D)-positive baby. Usually given no later than 72 hours postdelivery. If the Rh status of the infant is unknown at 72 hours, administer to the mother at that time. Should be given as soon as possible up to 28 days after delivery. This second dose postdelivery (first dose given predelivery; see above) can reduce treatment failure.
Rhophylac:
1,500 IU (300 mcg).
WinRho SDF:
600 IU (120 mcg).
Postabortion, amniocentesis (after 34 weeks’ gestation), or any other manipulation late in pregnancy (after 34 weeks’ gestation):
Confirm Rho(D) negative status of patient. May be given IV or IM. Administer immediately after abortion or procedure associated with increased risk of Rh isoimmunization. Must be given within 72 hours. One half of a dose (a mini-dose [IM product]) may be given if a pregnancy terminates before 13 weeks’ gestation, administration within 3 hours is preferred. According to the literature, this mini-dose can provide 100% effectiveness in preventing Rh immunization.
Rhophylac:
1,500 IU (300 mcg).
WinRho SDF:
600 IU (120 mcg).
Postamniocentesis before 34 weeks’ gestation or after chorionic villus sampling:
Confirm Rho(D) negative status of patient. May be given IV or IM.
Rhophylac or WinRho SDF:
1,500 IU (300 mcg) immediately after the procedure. Repeat WinRho SDF every 12 weeks during the pregnancy.
Threatened abortion:
Confirm Rho(D) negative status of patient. May be given IV or IM.
Rhophylac or WinRho SDF:
1,500 IU (300 mcg) as soon as possible.
Transfusion or fetal hemorrhage:
Confirm Rho(D) negative status of patient. May be given IV or IM. Administer within 72 hours of an incompatible event involving Rho(D) positive blood such as exposure to incompatible blood transfusions (Rh+ whole blood or Rh+ red blood cells) or massive fetal hemorrhage.
Rhophylac:
Give 100 IU (20 mcg) per each 2 mL transfused blood or per 1 mL erythrocyte concentrate. In cases of known or suspected excessive feto-maternal hemorrhage, the number of fetal red blood cells in the maternal circulation should be determined. If testing is not feasible and excessive feto-maternal hemorrhage cannot be excluded, administer a dose of 1,500 IU (300 mcg).
WinRho SDF:
Give up to 3,000 IU (600 mcg) every 8 hours IV or 6,000 IU (1,200 mcg) every 12 hours IM until the total dose is administered. Total IV dose is 45 IU (9 mcg) for every milliliter of Rh+ whole blood exposure or 90 IU (18 mcg) for every milliliter of Rh+ red blood cell exposure. Total IM dose is 60 IU (12 mcg) for every milliliter of Rh+ whole blood exposure or 120 IU (24 mcg) for every milliliter of Rh+ red blood cell exposure.
Treatment of immune thrombocytopenic purpura (ITP); adults and pediatric patients:
WinRho SDF:
Confirm Rho(D)-positive status of patient. Must be given IV. Hemoglobin should be greater than 10 Gm/dL. Give 250 IU/kg (50 mcg/kg) of body weight as the initial dose. May be given as a single dose or divided in half and given on two consecutive days. If response to the initial dose is adequate, maintenance doses of 125 to 300 IU/kg (25 to 60 mcg/kg) may be given. If response to the initial dose is inadequate, see Dose Adjustments. Dose and frequency based on patient’s clinical response (e.g., RBC, hemoglobin, reticulocyte levels, and platelet counts); see Dose Adjustments.
Pediatric dose
Treatment of ITP:
WinRho SDF:
See Usual Dose and Dose Adjustments.
Dose adjustments (international units [IU])
Pregnancy and obstetrical conditions:
WinRho SDF suggests protection must be maintained throughout pregnancy once Rho(D) immune globulin is administered. Level of passively acquired anti-Rho(D) should not fall below levels required to prevent an immune response to Rho(D)+ blood. Additional doses should be given every 12 weeks during pregnancy and at delivery unless the previous dose was administered within 3 weeks and there is less than 15 mL of fetomaternal red blood cell hemorrhage during delivery.
Suppression of Rh isoimmunization:
Rhophylac and WinRho SDF:
A large fetomaternal hemorrhage may cause an incorrect evaluation by standard tests of the amount of Rho(D) IGIV required. Assess the amount of hemorrhage and adjust dose accordingly.
Treatment of ITP: Adults and pediatric patients:
WinRho SDF:
If the hemoglobin level is less than 10 Gm/dL before or after the initial dose, reduce the initial dose and/or maintenance doses to 125 to 200 IU (25 to 40 mcg)/kg to minimize the risk of increasing the severity of anemia in the patient. ■ In patients with adequate platelet response to the initial dose, adjust maintenance doses based on platelet and hemoglobin levels. ■ If response to the initial dose is inadequate, adjust subsequent doses as follows:
Hemoglobin above 10 Gm/dL: redose with 250 to 300 IU (50 to 60 mcg)/kg.
Hemoglobin is 8 to 10 Gm/dL: redose with 125 to 200 IU (25 to 40 mcg)/kg.
Hemoglobin below 8 Gm/dL: use with caution; may increase severity of anemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


