22 Opioid intravenous anesthetics
Agonist: A drug with a specific cellular or receptor affinity that produces a predictable response.
Antagonist: A drug that exerts an opposite action to that of another or competes for the same receptor site or sites.
Breakthrough Pain: A transient increase in the intensity of pain from a baseline pain level that is no greater than moderate.
Dysphoria: A disorder of affect that is characterized by depression and anguish.
Endogenous: Originating inside the body.
Endorphins: These opioid peptides are produced in the body and are composed of many amino acid (protein) substances that attach to opioid receptors in the central nervous system and the peripheral nervous system for reduction of pain.
Exogenous: Originating outside the body.
Fixed Chest Syndrome: Rigidity of the diaphragmatic and intercostal muscles.
Mydriasis: Dilation of the pupil of the eye.
Opiate Receptor: Receptors that are transmembrane proteins that bind to endogenous opioid neuropeptides and exogenous morphine and similar compounds. They are designated mu, kappa, and delta subtypes of the opiate receptor.
Opioid: A drug that contains opium or a derivative of opium along with semisynthetic or synthetic drugs that have opium-like properties.
Piloerection: Erection of the hairs of the skin.
The immediate postanesthesia phase is when the patient is most vulnerable to complications (see Chapter 29).1 Many drugs that have residual anesthetic effects well into the postanesthesia period are used in modern anesthesia care. These agents include the potent inhaled agents, muscle relaxants, benzodiazepines, and opioids. Respiratory depression is the most common adverse event in the postanesthesia care unit (PACU); therefore the use and understanding of the various opioid agents optimize patient outcomes. In addition, the reduction of pain in the PACU is one of the primary focuses of care in the perianesthesia phase. In addition to assessing pain, the perianesthesia nurse must take into the evaluation of pain the preoperative, intraoperative, and postanesthesia phase of the surgical patient. Because all pain-reducing drugs must be taken into account during the pain assessment in the PACU, the mixed action or agonist-antagonist combination drugs are presented in this chapter. With a detailed description of the major opioids used in the perianesthesia period provided, the PACU nurse will be able to make excellent informed decisions in regard to the anticipated outcome of the patient of pain reduction and comfort.
Concept of opioids and opioid receptors
Opioids are the substances, either natural or synthetic, that are administered into the body (exogenous) and bind to specific receptors to produce a morphine-like or opioid agonist effect. The endogenous opioids are the endorphins. The endorphins, which are produced in the body, attach to the opioid receptors in the central nervous system (CNS) to activate the body’s pain modulating system. The term opioid is used because of the multitude of synthetic drugs with morphine-like actions; with the advent of receptor physiology, opioid has replaced the term narcotic, which is derived from the Greek word for “stupor” and usually refers to both the production of the morphine-like effects and the physical dependence.2
The naturally occurring alkaloids of opium are divided into two classes: phenanthrene and benzylisoquinoline. The principal phenanthrene series of drugs includes morphine, codeine, and thebaine. Papaverine and noscapine, which lack opioid activity, represent the benzylisoquinoline alkaloids of opium.3
The synthetic opioids have been produced with the modification of the chemical structure of the phenanthrene class of drugs. Drugs such as fentanyl (Sublimaze) and meperidine (Demerol) are examples of synthetic opioids.2,4
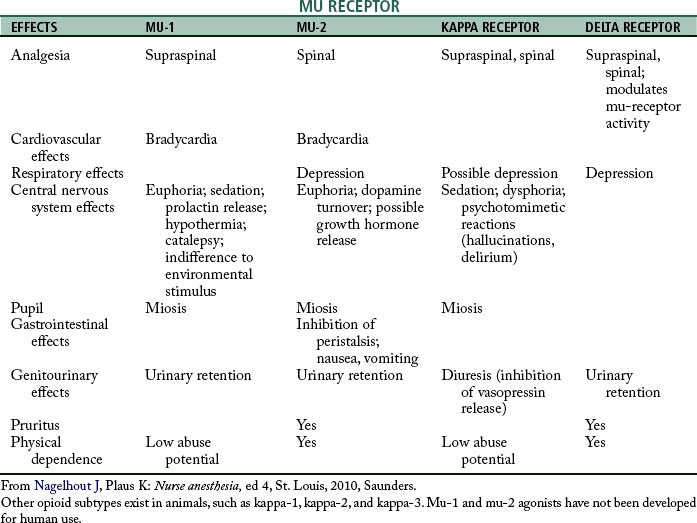
The identification of specific opioid receptors has enhanced the understanding of the agonist and antagonist actions of this category of drugs. The opioid receptors are located in the CNS, principally in the brain stem and spinal cord. These receptors have been determined by the pharmacologic effect they produce when stimulated by a specific agonist along with how the effect is blocked by a specific antagonist. The three major categories of opioid receptors are the mu (μ), delta (δ), and kappa (κ).4,5
The mu receptors are mainly responsible for the production of supraspinal analgesia effects with stimulation. These receptors are further divided into mu-1 and mu-2 types. Activation of the mu-1 receptors results in analgesia; when the mu-2 receptors are stimulated, hypoventilation, bradycardia, physical dependence, euphoria, and ileus can result. The mu receptors are activated by morphine, fentanyl, and meperidine. The drug that is specific to the mu-1 receptor is meptazinol, which is supraspinal in regard to analgesia; the mu-2 receptor analgesia occurs at the spinal level. Other characteristics of the mu-1 and mu-2 receptors are summarized in Table 22-1. Stimulation of the kappa receptors results in spinal analgesia, dysphoria, hallucinations, hypertonia, tachycardia, tachypnea, mydriasis, sedation, and miosis, with little effect on ventilation. The drugs that possess both opioid agonist and antagonist activities, such as nalbuphine (Nubain), have their principal action on the kappa opioid receptors. The delta opioid receptors, when stimulated, serve to modulate the activity of the mu receptors and cause depression and urinary retention. The drug naloxone (Narcan) attaches to all the opioid receptors and thus serves as an antagonist to all the opioid agonists (see Table 22-1).2,4
Opioids
The administration of opioids in the perioperative period is not without the concern of overdose. The major signs of overdose with opioids are miosis, hypoventilation, and coma. If the patient becomes severely hypoxemic, mydriasis can occur. Airway obstruction is a strong possibility because the skeletal muscles become flaccid. Hypotension and seizures can also occur. The treatment for an opioid overdose is mechanical ventilation and the slow titration of naloxone. Consideration must always be given to the fact that some patients who become overdosed with an opioid may indeed be already physically dependent. Naloxone can precipitate an acute withdrawal syndrome.2
Meperidine hydrochloride
Meperidine (Demerol) was discovered in 1939 by Eisleb and Schauman. Because it is chemically similar to atropine, it was originally introduced as an antispasmodic agent and was not used as an opioid anesthetic agent until 1947. The main action of this drug is similar to morphine; it stimulates the subcortical mu receptors, which results in an analgesic effect. Meperidine is approximately one tenth as potent as morphine and has a duration of action of 2 to 4 hours. The onset of analgesia is prompt (10 minutes) after subcutaneous or intramuscular administration. All pain, especially visceral, gastrointestinal, and urinary tract, is satisfactorily relieved. This drug causes less biliary tract spasm than morphine; however, in comparison with codeine, meperidine causes greater biliary tract spasm. It produces some sleepiness but causes little euphoria or amnesia. Meperidine increases the sensitivity of the labyrinthine apparatus of the ear, which explains the dizziness, nausea, and vomiting that sometimes occur in ambulatory patients.2,4
Meperidine is generally metabolized in the liver; less than 5% is excreted unchanged by the kidneys. However, because of a toxic metabolite of meperidine, patients who are administered this drug may have seizures.2 Meperidine is partially metabolized to normeperidine which has some analgesic effects, but more importantly, lowers the seizure threshold and can induce CNS excitability. Meperidine probably should not be administered to elderly patients because renal dysfunction may occur and less tolerance to normeperidine.
Because of its spasmolytic effect, meperidine is the drug of choice for biliary duct, distal colon, and rectal surgery. It offers the advantages of little interference with the physiologic compensatory mechanisms, low toxicity, smooth and rapid recovery, prolonged postoperative analgesia, excellent cardiac stability in patients at poor risk, and ease of detoxification and excretion.2,4,5 Meperidine is used most often with procedures now, and not typically for long-term pain management because of the effects of normeperidine over time.
Morphine
Morphine, one of the oldest known drugs, has only recently been used as an opioid intravenous anesthetic agent. Alkaloid morphine is from the phenanthrene class of opium. The exact mechanism of action of morphine is unknown. In humans, it produces analgesia, drowsiness, changes in mood, and mental clouding. The analgesic effect can become profound before the other effects are severe and can persist after many of the side effects have almost disappeared. With direct effect on the respiratory center, morphine depresses respiratory rate, tidal volume, and minute volume. Maximal respiratory depression occurs within 7 minutes after intravenous injection of the drug and 30 minutes after intramuscular administration. After therapeutic doses of morphine, the sensitivity of the respiratory center begins to return to normal in 2 or 3 hours, but the minute volume does not return to preinjection level until 4 or 5 hours have passed.
Morphine can cause nausea and vomiting, especially in ambulatory patients, because of direct stimulation of the chemoreceptor trigger zone. The emetic effect of morphine can be counteracted with opioid antagonists and phenothiazine derivatives such as prochlorperazine (Compazine), dexmedetomidine (Precedex), or the 5-HT3 receptor antagonist ondansetron (Zofran). Histamine release has been noted with morphine, and morphine also causes profound constriction of the pupils, stimulation of the visceral smooth muscles, and spasm of the sphincter of Oddi.5–7
Morphine is detoxified by conjugation with glucuronic acid. Ninety percent is excreted by the kidneys, and 7% to 10% is excreted in the feces via the bile.2
Methadone
The actions of this synthetic opioid agonist resemble morphine; side effects include depression of ventilation, miosis, constipation, and biliary tract spasm. Clinically, the sedative and euphoric actions of methadone appear to be less than those produced by morphine.5
Hydromorphone
Hydromorphone (Dilaudid), which is a derivative of morphine, was developed in Germany in the 1920s and released to the mass market in the late 1920s. The drug has a renewed popularity for the PACU and can be administered intravenously, intramuscularly, rectally, or orally. The drug profile in regard to its analgesia and side effects is similar to morphine. Hydromorphone is recommended for patients in renal failure because of its virtual lack of active metabolites after its breakdown in the liver. It has a high solubility and a rapid onset of action and appears to have less troublesome side effects and dependence liability profile as compared with morphine. Because of its high lipid solubility, hydromorphone can be administered via epidural or spinal for a wide area of anesthesia as compared with duramophine.2
Hydromorphone, like all opioids, is a CNS depressant and has actions and side effects similar to morphine. Its depressant effects can be enhanced with beta-blockers and alcohol. The duration of action of this drug is approximately 2 hours, with a peak action in approximately 30 minutes with intravenous administration.
Fentanyl
Janssen and colleagues8 introduced a series of highly potent meperidine derivatives that were found to render the patient free of pain without affecting certain areas in the CNS. Fentanyl (Sublimaze) appeared to be of special interest. In regard to analgesic properties, fentanyl is approximately 80- to 125-fold more potent as morphine and has a rapid onset of action of 5 to 6 minutes and a peak effect within 5 to 15 minutes. The analgesia lasts 20 to 40 minutes when administered intravenously. Via the intramuscular route, the onset of action is 7 to 15 minutes; the analgesia usually lasts 1 to 2 hours. When fentanyl is administered as a single bolus, 75% of the drug undergoes first-pass pulmonary uptake. That is, the lungs serve as a large storage site and this nonrespiratory function of the lung (see Chapter 12) limits the amount of fentanyl that actually reaches the systemic circulation. If the patient receives multiple doses of fentanyl via single injections or infusion, the first-pass pulmonary uptake mechanism becomes saturated and the patient has a prolonged emergence because of increased duration of the drug. Consequently, during the admission of the patient to the PACU, the postanesthesia nurse must determine the frequency and amount of intraoperative fentanyl administration. Patients who have received a significant amount of fentanyl via infusion or via titration should be continuously monitored for persistent or recurrent respiratory depression. In addition, fentanyl has been implicated in what is called a delayed-onset respiratory depression. In some patients, a secondary peak of the drug concentration in the plasma occurs approximately 45 minutes after the apparent recovery from the drug. This syndrome can occur when some of the fentanyl becomes sequestered in the gastric fluid and then can become recycled into the plasma in approximately 45 minutes. Therefore, in the PACU, all patients who have received fentanyl should be continuously monitored for respiratory depression for at least 1 hour from the time of admission to the unit.2
Fentanyl can be administered during surgery at three different dose ranges, depending on the type of surgery and the desired effect. For example, the low-dose range of 2 to 20 mcg/kg attenuates moderately stressful stimuli. The moderate dose range is 20 to 50 mcg/kg and strongly obtunds the stress response. The megadose range of as much as 150 mcg/kg blocks the stress response and is particularly valuable when protection of the myocardium is critical.4
Fentanyl can be used alone in a nitrous-opioid technique. It also is used in the PACU in the form of a low-dose intravenous drip for pain relief; however, fentanyl is usually given slowly intravenously in the PACU for breakthrough pain (see Table 22-2 for helpful calculation of milligram to microgram dosage information).
Table 22-2 Example of Conversion of Dosage Calculations from Milligrams to Micrograms
| MILLIGRAMS | MICROGRAMS | MILLILITERS OF FENTANYL |
|---|---|---|
| 0.025 | 25 | 0.5 |
| 0.05 | 50 | 1.0 |
| 0.10 | 100 | 2.0 |
| 0.15 | 150 | 3.0 |
| 0.20 | 200 | 4.0 |
| 0.25 | 250 | 4.5 |
| 0.50 | 500 | 10.0 |
| 1.00 | 1000 | 20.0 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


