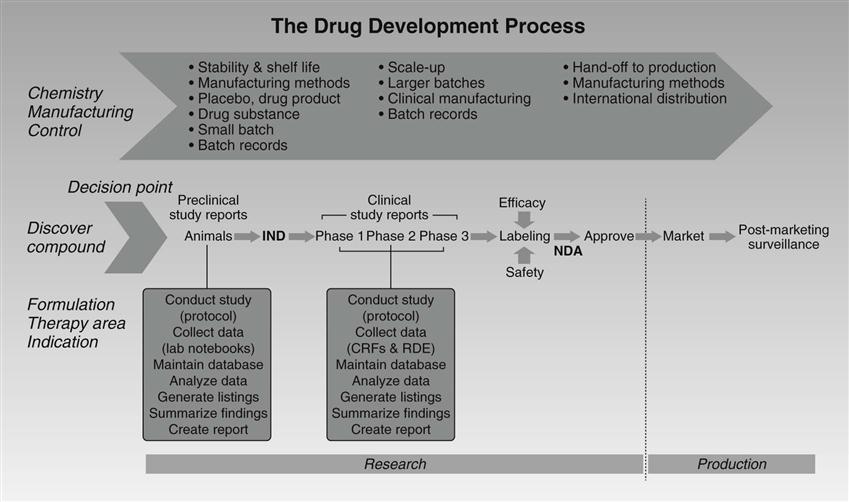
Ruth Merkatz and Elyse I. Summers “Get to the table and be a player, or someone who does not understand nursing will do that for you.” —Loretta Ford, EdD, RN, PNP, FAAN, FAANP Americans take a lot of medicine. Pharmaceutical drugs are available because of a robust research infrastructure that works in tandem with the United States Food and Drug Administration’s (FDA’s) review and approval process and allows for new, innovative treatments to become available to a populace clamoring for cures for life-threatening or chronic illnesses. As this chapter explains, with so many drugs being used by so many people, it is of paramount importance that the safety and efficacy of these products be demonstrated and monitored over time. Nurses help assure the safety and welfare of research participants and patients during development, early-phase use, and the postapproval monitoring periods of these life-enhancing and life-saving products. For decades, Americans have been enthusiastic consumers of the thousands1 of drugs approved for use in this country. And, as we moved from the late twentieth century into the first decade of the twenty-first century, U.S. prescription drug use continued to grow. The Centers for Disease Control and Prevention’s (CDC’s) National Center for Health Statistics (NCHS) National Health and Nutrition Examination Survey (NHNES) surveyed individuals regarding their “prescription drug use in the past month,” during the years 1988 to 1994. In this survey, almost 40% of the American populace had used a prescription drug in the course of a month’s time (NHNES, 2008). By the agency’s most recent survey, 2001 to 2004, the figure had grown to close to 50% (NHNES, 2008). Regardless of the survey time frame, the most significant factor in prescription drug usage is age. From 1988 to 1994, almost 74% of individuals 65 years and older reported drug use (NHNES, 2008); from 2001 to 2004, it was 87% for this same cohort (NHNES, 2008). Moreover, between 1988 and 1994 and between 2001 and 2004, the elder group increased the number of medications they were using. Just over 35% of this cohort reported using three or more prescription drugs in the earlier time frame, a figure that rose to almost 60% in the early 2000s (NHNES, 2008). The trend in U.S. medication is clear. In any given week, a solid majority of the population (81% of adults) will take at least one prescription or non-prescription medication; many will use at least one prescription drug (50% of adults), and some will take five or more (7% of adults) (Kaufman et al., 2002). For drugs to be available to American consumers, they are to be safe and effective. However, this was not always the case. The central role the FDA now occupies on the bench-to-bedside continuum took years to establish and is, in some ways, ever-evolving. The oldest comprehensive consumer protection agency in the U.S. federal government, the FDA’s activities trace back to the mid-1800s when a section within the Patent Office began carrying out chemical analyses of agricultural products (U.S. Food and Drug Administration [FDA], n.d.-a). The 1906 Wiley Act was the first U.S. law to prohibit interstate commerce of adulterated and misbranded food and drugs. However, it wasn’t until after the 1937 sulfanilamide tragedy that the FDA gained meaningful authority to ensure drug safety. Elixir of Sulfanilamide, marketed as a new sulfa wonder drug that children could ingest easily, was actually prepared with a highly toxic chemical analogue of anti-freeze that led to the deaths of more than 100 children (Ballentine, 1981). The result of this tragedy and subsequent public exposure was the Food, Drug, and Cosmetic Act of 1938. This law mandated premarket approval of all new drugs based on a demonstration of safety to the FDA; it authorized FDA inspection of manufacturing facilities; and, it granted FDA authority over cosmetics and medical devices. In 1962, the thalidomide disaster, centered in Europe, prompted the expansion of the FDA’s regulatory purview to also include “effectiveness.” Outside the U.S., thalidomide had been marketed as another “wonder drug,” but its use by pregnant women resulted in the birth of thousands of severely deformed babies (phocomelia defect). Alarmed by the European experience, U.S. Senator Estes Kefauver held hearings on the American drug approval process, pushed for, and ultimately secured passage of the Kefauver-Harris Amendments (FDA, n.d.-b). The Kefauver-Harris Amendments required the FDA to assess the efficacy of all drugs introduced since 1938; instituted stricter agency control over drug trials (including formal introduction of the concept of and requirement for patient informed consent in drug trials); transferred to the FDA from the Federal Trade Commission regulation of prescription drug advertising; established good manufacturing practices as a drug industry requirement; and granted the FDA greater powers of inspection and enforcement. Since the 1962 amendments, new drug development has become an increasingly costly, time-consuming endeavor (DeMasi, Hansen, & Grabowski, 2003). Estimates on the amount of time it now takes for a new compound to reach approval are as high as 12 to 13 years (Figure 28-1). Generally speaking, new drug discovery and development proceeds in a sequence of phases, sometimes overlapping (DeMasi et al., 2003). Prior to any human testing, a new drug’s sponsor, typically a pharmaceutical company, but sometimes, a non-governmental organization or government agency, tests a new compound in assays and animal models. If encouraged by the results, the sponsor submits to the FDA an investigational new drug application (IND) that contains all known information about the compound, including manufacturing information to demonstrate that the test article can be produced consistently with high quality. The IND also specifies the clinical research plan and protocol for phase 1 studies. Unless the FDA affirmatively objects, the IND is automatically allowed after 30 days, and human clinical trials can begin. In accordance with federal regulations, local institutional review boards (IRBs) charged with protecting research participants must approve these trials (FDA Protection of Human Subjects Regulations, 1980; FDA Institutional Review Board Regulations, 1981). IRBs review protocols relative to their scientific merit, risks and benefits, and procedures for gaining informed consent. They are also expected to review plans for recruiting volunteers to avert inappropriate incentives or coercive tactics (FDA Protection of Human Subjects Regulations, 1980; FDA Institutional Review Board Regulations, 1981). A drug’s premarket clinical testing must proceed through three successive phases consistent with the required safety and efficacy standards. In Phase 1, a small number of usually healthy volunteers are given the drug to establish safe dosages, and characterize the absorption, distribution, metabolic effects, and excretion (ADME) of the drug. These data help determine the drug’s pharmacokinetic (PK) profile (the way the body processes a drug), its tolerability and safety profile, and pharmacodynamics (PD), the way the drug works in the body. Phase 2 trials include a few hundred volunteers who have the targeted disease or condition to determine an effective dose and to begin to evaluate efficacy and short-term risks. In Phase 3, studies are conducted to evaluate safety and efficacy in several hundred to several thousand patients or volunteers at risk for the drug’s targeted condition (e.g. a new female contraceptive is tested in women at risk for pregnancy; a colon cancer treatment is tested in people with this condition). Depending on a drug’s indication, testing occurs in hospitals and/or outpatient settings. Phase 3 studies are usually randomized, controlled multicenter trials with results of the new drug tested against an approved standard therapeutic agent (sometimes referred to as the “gold standard”). The goal of these studies is to gather precise information on a drug’s effectiveness for specific indications, determine whether or not the drug produces a broader range of adverse events (AEs) than those exhibited in the smaller Phase 1 and 2 study populations, and identify the best way to administer and use the drug for its intended purpose. These studies also can uncover infrequent side effects. Once a sponsor has finished preclinical and clinical testing, analyzed the data, and written reports in accordance with regulatory guidelines, it can compile and submit a new drug application (NDA) to the FDA, which will assess it for completeness before filing and initiating its review. In addition to the aforementioned components, the NDA must also include detailed chemistry, manufacturing and quality controls (CMC) documentation to ensure that the marketed product will adhere to consistent safety and purity standards. In accordance with a time frame mandated by the Prescription Drug User Fee Act (PDUFA) that took effect initially in 1993, the FDA must complete its review in a timely fashion. The approval process for most new drugs takes 12 months on average, while priority applications are generally reviewed within 6 months. During the review period, the FDA may seek clarification of data contained in the NDA or ask for new information. To augment its internal review, the FDA sometimes will convene an advisory “expert” panel to discuss the NDA publicly and advise the agency regarding the new drug’s approvability. These panels are comprised of health professionals with expertise in the therapeutic area applicable to the drug under review, including statisticians and/or epidemiologists, and generally a consumer and industry representative. Historically and currently, physicians compose the majority of health professional representatives to these panels. On occasion, nurses are invited to serve, but often as consumer representatives who may or may not be voting members. As part of the approval process, information gleaned during the phases of drug testing forms the basis for product labeling information. The FDA’s guidance on drug labeling ensures that package inserts include the same types of information in a standardized format, although the titles may vary according to manufacturer preference. Developing this label is a lengthy process that often includes heated negotiation between sponsor representatives and FDA staff regarding the exact wording of an approved drug label. Every single word is important. Adding an indication or removing a contraindication can have a several-million-, even billion-dollar impact for a sponsor as well as significant health consequences for the medication-taking public. The addition of a “black box” warning (the strongest warning the FDA issues in its drug labeling) on antidepressants prescribed to children and adolescents, for example, not only reduced prescriptions for selective serotonin reuptake inhibitors (SSRIs) by an estimated 22% in the U.S., but also was associated with reducing aggregate rates of diagnosis and treatment of pediatric depression (Gibbons et al., 2007; Libby et al., 2007). A label’s contents, therefore, are of paramount importance to nurses who care for patients and/or are responsible for updating other nurses on labeling changes. Participants in clinical trials prior to marketing must meet strict inclusion/exclusion criteria to minimize safety risks and to increase the likelihood of achieving reliable results. Consequently, clinical trial populations may not necessarily represent typical users of drugs once approved and released into the marketplace. Eligibility criteria for clinical trial volunteers, for example, often require confirmation of normal kidney and liver function as measured through serum chemistries such as creatine clearance, blood urea nitrogen, SGOT (AST), and SGPT (ALT). Abnormalities of these functions can limit participation, especially among older adults and those with chronic conditions who may fail to meet eligibility criteria. Given that most drugs are metabolized through the liver or kidneys, which affects drug metabolism and excretion and influences concentration of drugs in the tissues, eliminating such groups during clinical trials can result in biased or incomplete results relative to both safety and efficacy, and may mask toxicities (Benet, Massoud & Gambertoglio, 1984; Rowland & Tozer, 1995). Also, the relative size of trials included in the development process is small (e.g., 300 to 3000 volunteers) compared to the much larger number of individuals who will take a drug once available—another factor that can limit wide generalizations of clinical trial results. And due to the limited duration of trials, data from volunteer usage of a new drug generally cannot reveal long-term effects. Once marketed, a new drug may also be prescribed for unapproved uses not specified on the drug label and/or prescribed along with other medications that can alter the known PK and PD. While sponsors typically study the way a new drug interacts with certain other types of drugs, it is impossible to identify all potential interactions. Ultimately, all of these scenarios compound risks that could not have been identified easily during the time-sensitive drug-approval process, but then emerge post-marketing. That is why evidence is accumulating for vigilant post-marketing surveillance and why a number of drugs either have received strict safety warnings after approval, including the black box warning, or been withdrawn from the market (Table 28-1). TABLE 28-1 Chronology of Significant U.S. Drug Withdrawals (2000-2009) Source: www.fda.gov.
Nursing’s Influence on Drug Development and Safety
Historical Background
Current Drug Approval Framework
Postmarketing Surveillance
Year
Drug
Reason for Withdrawal
2009
Efalizumab (Raptiva)
Increased risk of progressive multifocal leukoencephalopathy
2008
Aprotinin (Trasylol)
Increased risk of death
2007
Tegaserod (Zelnorm)
Increased risk of serious cardiovascular adverse events
2007
Pergolide (Permax)
Risk of heart valve damage
2006
Gatifloxacin (Tequin)
Liver damage
2005
Natalizumab (Tysabri)
Progressive multifocal leukoencephalopathy
2005
Pemoline (Cylert)
Liver toxicity
2005
Hydromorphone extended-release (Palladone)
High risk of accidental overdose when taken with alcohol
2004
Rofecoxib (Vioxx)
Increased risk of CV events, including MI and stroke
2001
Rapacuronium (Raplon)
Risk of fatal bronchospasm; unexplained fatalities
2001
Cerivastatin (Baycol)
Risk of fatal rhabdomyolysis
2000
Phenylpropanolamine (i.e., Dexatrim)
Risk of hemorrhagic stroke
2000
Cisapride (Propulsid)
Risk of serious cardiac arrhythmias and death
2000
Alosetron (Lotronex)
Risk of fatal complications of constipation
2000
Troglitazone (Rezulin)
Risk of irreversible liver damage and liver failure
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Nurse Key
Fastest Nurse Insight Engine
Get Clinical Tree app for offline access
