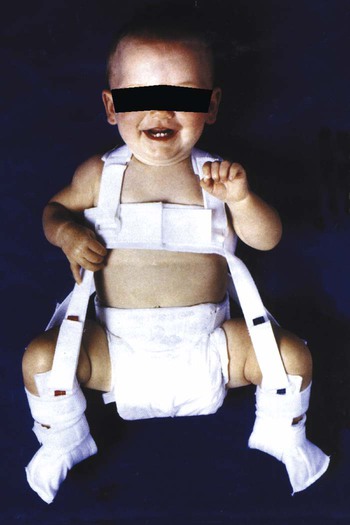
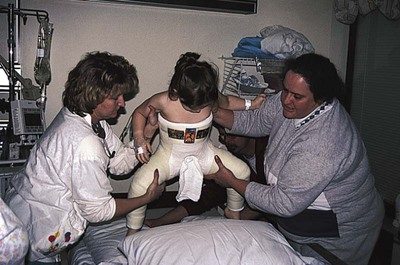
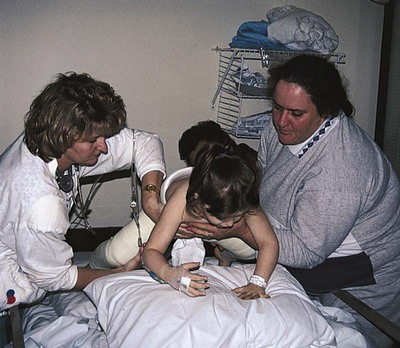
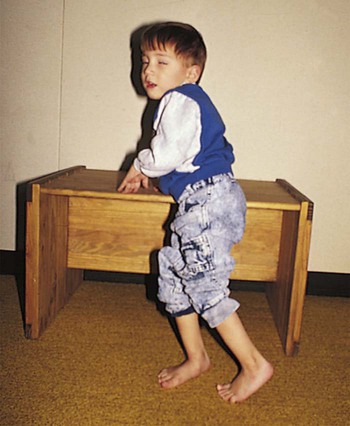
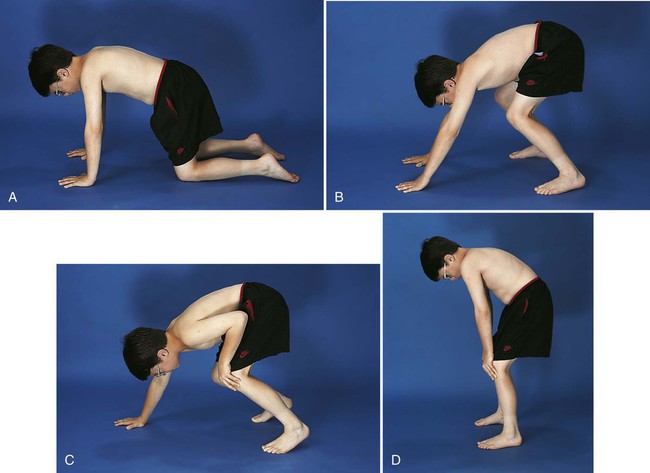
1. Define the vocabulary terms listed 2. Describe the musculoskeletal changes that occur from infancy 3. Identify two ways in which the bones of a toddler differ from those of an adult 4. List the home instruction needed for a infant in a Pavik harness 5. Describe the nursing care of a child with Duchenne muscular dystrophy 6. Discuss the nursing measures for a child in a body cast 7. List various types of pediatric fractures 8. Identify the pathophysiology of Legg-Calvé-Perthes disease and the management approach 9. Formulate a nursing care plan for the adolescent confined in a brace for the treatment of scoliosis 10. Describe several measures designed to prevent sports injury Clubfoot, one of the most common congenital deformities of the skeletal system, is characterized by a foot that has been twisted inward or outward or toes higher or lower than the heel (Figure 17-1). These deformities can vary from mild and flexible to severe and rigid. The incidence rate is about 1 in 1000 births, with boys affected twice as often as girls. Several variations are recognized. Talipes (talus, heel; pes, foot) equinovarus (equinus, extension; varus, bent inward) is seen in 95% of cases. The feet are turned inward, and the untreated child walks on the balls of the foot and the outer borders of the feet. Bilateral clubfeet occur in 50% of cases. While the exact cause is unknown, recent investigations suggest a genetic influence (Horn and Davidson, 2010). Clubfoot may occur in children with congenital and chromosomal syndromes such as cerebral palsy, spina bifida, and myelomeningocele. Idiopathic clubfoot is thought to result from an unrecognized neuromuscular disorder. Developmental dysplasia of the hip (DDH) is a common orthopedic deformity. The term hip dysplasia is a broad description applied to various degrees of deformity that may involve subluxation or dislocation and may be either partial or complete. The head of the femur is partially or completely displaced from a shallow hip socket (acetabulum). Both hereditary and environmental factors appear to be involved in the cause (Box 17-1). Hip malformation, joint laxity, breech position, and race may all contribute. DDH is seven times more common in girls than in boys. Newborn infants seldom have complete dislocation. When the infant begins to walk, the pressure exerted on the hip can cause a complete dislocation. Accordingly, early detection and treatment are of particular importance in this condition. Subluxation of the hip is commonly discovered at the time of the newborn examination. Ongoing screening for DDH should be done during routine health examinations during the first year of life. One of the most reliable signs is a limitation of abduction of the leg on the affected side. When the infant is placed on the back with knees and hips flexed, the physician can press the femur related to the normal hip back until it almost touches the examining table. On the affected side, however, this can be accomplished only partially. Also, the knee on the side of the dislocation appears to be shorter and the skin folds of the thigh are often asymmetrical. When the infant is prone, one hip is higher than the other (Figure 17-2). In some infants younger than 4 weeks of age, the physician can actually feel and hear the femoral head slip into the acetabulum under gentle pressure. This is called Ortolani’s sign or Ortolani’s click and is considered diagnostic. If the child has begun walking and has had no treatment, he or she will display the characteristic waddling gait and will test positive for the Trendelenburg gait test (pelvic drops on the side of the affected hip joint when standing on one leg). Bilateral dislocation may occur; however, unilateral dislocation is more common. Radiographs and ultrasound scans confirm the diagnosis. From birth to approximately 6 months of age, abduction of the hips is maintained with the use of the Pavlik harness (Figure 17-3). The harness is worn full time until stability is attained plus 2 months, then a decrease in wearing time begins. Weaning time from the harness is gradual until normal hip function is established by ultrasound or x-ray. The Pavlik harness allows the infant to move the legs. If the dislocation is severe or has not been detected until the child has begun to walk, it may be necessary to use traction. This pulls the head of the femur down to the correct position opposite the acetabulum. After the traction has stretched the muscles enough to allow the hip to be placed in the acetabulum, the dislocation is reduced with general anesthesia and a spica cast is applied to hold the abduction. This type of cast is shown in Figure 17-4. The length of time that the child remains in the cast varies according to progress, growth, and the condition of the cast; however, it is usually from 3 to 4 months. During this time, the cast may be changed about every 6 weeks. Sometimes surgery is necessary. In this case, open reduction of the dislocation or repair of the shelf of the hip bone is done. A cast is applied after surgery to keep the femur in the correct position. After removal of the spica cast, some children may require an abduction brace. The brace is worn for 4 to 8 weeks, and then only at nighttime for 1 to 2 years (Canale and Beaty, 2008). For children 6 to 18 months of age, the Pavik harness is less effective and traction is required. The body spica cast encircles the waist and extends to the ankles or toes. General cast care should be reviewed (see Cast Care section under Clubfoot in this chapter). Skill 17-1 describes how to turn and position a child in a body cast. Skill 17-1 Turning and Positioning a Child in a Body Cast 1. Use two people to turn a child in a body cast. 2. Lift the child and place in the prone position. 3. Do not use crossbars between the legs as handles. 4. With the child in the prone position, place a pillow under the chest and under each leg to prevent pressure on the toes. 5. With a bedpan, support the upper back and legs with pillows so that body alignment is maintained. Cerebral palsy (CP) refers to a group of nonprogressive disorders that affect the motor centers of the brain, causing problems with movement and coordination. Often children with CP have associated language, perceptual, and intellectual deficits. It is one of the most common disabling conditions seen in children, occurring in approximately 2.5 per 1000 live births (Dodge, 2008). • Congenital problems involving the central nervous system (e.g., hydrocephalus, hemorrhage) • Postnatal trauma (head injuries) or infections (meningitis, encephalitis) • Conditions of pregnancy or labor that interfere with oxygen reaching the fetal brain (e.g., premature separation of the placenta, prolonged labor) • Exposure during pregnancy to infections (e.g., German measles or rubella, cytomegalovirus, toxoplasmosis) or toxins The symptoms of CP vary with each child and may range from mild to severe (see Did You Know?). About two thirds of children who have CP are intellectually impaired (National Institute of Neurological Disorders and Stroke, Cerebral Palsy: Hope Through Research, 2010). CP is suspected during infancy when developmental milestones are not met. Diagnostic tests include MRI, electroencephalography, computed tomography (CT), and screening for metabolic disorders. Brain tumors must also be ruled out. Early recognition is important so that early intervention can begin. For the Child with Cerebral Palsy • Seizures, usually within 48 hours of birth • Delay in reaching developmental milestones such as sitting, crawling, creeping, standing, and reaching for objects • Difficulty with fine-motor skills such as holding feeding utensils, writing, and using scissors • Feeding difficulties such as poor sucking and swallowing, drooling, and persistent tongue thrust • Involuntary movements such as uncontrolled writhing motions of the hands • Increased muscle tone: Infant may be rigid when pulled to a sitting position; infant reflexes do not disappear at the normal time Children may have symptoms of more than one type of CP. Spastic CP is characterized by tension in certain muscle groups. When the child tries to move the voluntary muscles, jerky motions result, and eating, walking, and other coordinated movements are difficult to accomplish. The lower extremities are usually involved. The legs cross and the toes point inward (scissoring). Toe walking can occur from muscle tightness (Figure 17-5). Upper extremities, or upper and lower extremities on only one side of the body, can be affected. With athetoid or dyskinetic CP, the child has uncontrolled, slow, writhing movements that can increase during periods of emotional stress and disappear during sleep. These abnormal movements usually affect the hands, feet, arms, or legs and, in some cases, the muscles of the face and tongue, causing grimacing or drooling. Problems can also occur with the coordination of muscle movements needed for speech (dysarthria). Ataxia, or lack of muscle coordination, can be shown by disturbances of balance and depth perception. A wide-based gait accompanied by unsteadiness is generally present. Intention tremor may also be observed. As the child begins a voluntary movement, such as reaching for a book, there is a resultant trembling that affects the body part being used and worsens as the individual gets nearer to the desired object. Mixed CP is usually a combination of spastic and athetoid movements; however, other combinations of symptoms are possible. Medications may improve overall function in children with CP. Dantrolene reduces calcium release, thereby decreasing excitation-contraction in skeletal muscle. Skeletal muscle relaxants such as baclofen may decrease spasticity. Intrathecal baclofen delivered via a pump reservoir can help with spastic quadriparesis. Botulinum toxin type A (Botox) has also been successfully used to reduce spasticity (Burg et al., 2006). Antianxiety agents may relieve excessive motion and tension, especially in the athetoid child. Anticonvulsants are prescribed for children with seizures. The onset is generally between 2 and 6 years of age; however, a history of delayed motor development during infancy may be evidenced. A waddling gait, slowness in running or climbing, and enlarged rubbery muscles are indicative of this disorder. The calf muscles, in particular, develop pseudohypertrophy from the accumulation of fat and connective tissue. Other signs include frequent falling, clumsiness, contractures of the ankles and hips, and the Gower maneuver, a characteristic way of rising from the floor (Figure 17-6). Nurses function as team members along with personnel from many other disciplines in the care of the child with muscular dystrophy. The child should be encouraged to be as active as possible to delay muscle atrophy. Swimming and other activities that promote ROM and mobility for as long as possible are helpful. Nurses provide support for the many daily issues that occur by referring parents to other parents, camp programs, respite care, the Muscular Dystrophy Association (www.mdausa.org), public health nurses, home health agencies, family therapists, and eventually hospice care. Although there is no cure at present, different drug therapies, genetic engineering, and stem cell research are beginning to show promise and continue to be investigated (Quintero et al., 2009).
Musculoskeletal Disorders
 http://evolve.elsevier.com/Price/pediatric/
http://evolve.elsevier.com/Price/pediatric/
Musculoskeletal System
Clubfoot
Developmental Dysplasia of the Hip
Signs and Symptoms
Treatment
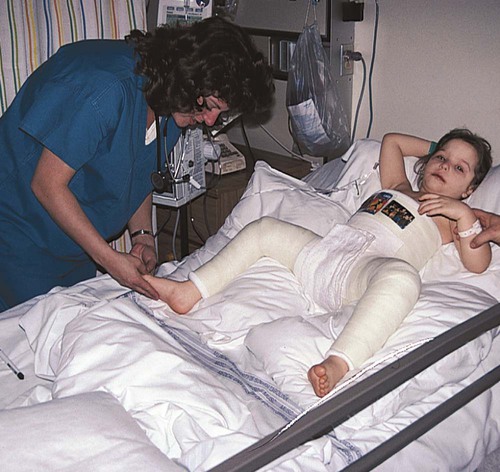
Nursing Care








Cerebral Palsy
Signs and Symptoms
 Did You Know?
Did You Know?
Treatment and Nursing Care
Duchenne Muscular Dystrophy
Signs and Symptoms
Treatment and Nursing Care
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Musculoskeletal Disorders
 ; p. 324)
; p. 324)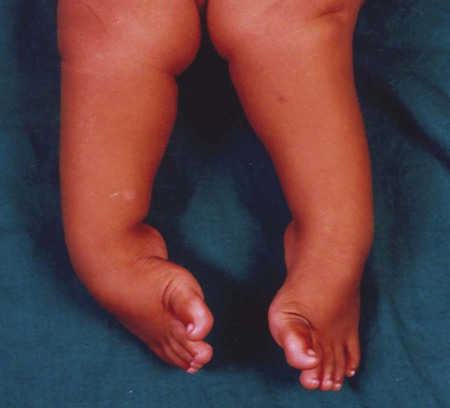
 Nursing Brief
Nursing Brief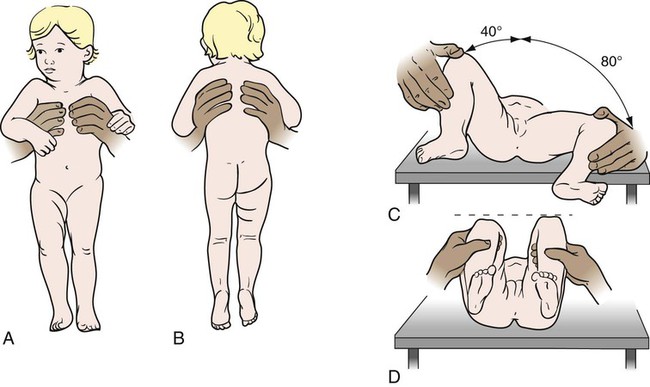
Get Clinical Tree app for offline access
