CHAPTER 8 Drug sensitivity varies with age. Infants are especially sensitive to drugs, as are the elderly. In the very young, heightened drug sensitivity is the result of organ immaturity. In the elderly, heightened sensitivity results largely from organ degeneration. Other factors that affect sensitivity in the elderly are increased severity of illness, the presence of multiple pathologies, and treatment with multiple drugs. The clinical challenge created by heightened drug sensitivity in the very young and in the elderly is the subject of Chapters 10 and 11, respectively. The impact of kidney disease is illustrated in Figure 8–1, which shows the decline in plasma levels of kanamycin (an antibiotic) following injection into two patients, one with healthy kidneys and one with renal failure. (Elimination of kanamycin is exclusively renal.) As indicated, kanamycin levels fall off rapidly in the patient with good kidney function. In this patient, the drug’s half-life is brief—only 1.5 hours. In contrast, drug levels decline very slowly in the patient with renal failure. Because of kidney disease, the half-life of kanamycin has increased by nearly 17-fold—from 1.5 hours to 25 hours. Under these conditions, if dosage is not reduced, kanamycin will quickly accumulate to dangerous levels. By altering pH partitioning (see Chapter 4), changes in acid-base status can alter the absorption, distribution, metabolism, and excretion of drugs. Figure 8–2 illustrates the impact of altered acid-base status on drug distribution. Specifically, it shows the results of altered acid-base status on the distribution of phenobarbital (a weak acid) in a dog. The upper curve shows plasma levels of phenobarbital. The lower curve shows plasma pH. Acid-base status was altered by having the dog inhale a mixture of gas rich in carbon dioxide (CO2), thereby causing respiratory acidosis. In the figure, acidosis is indicated by the drop in plasma pH. Note that the decline in pH is associated with a parallel drop in levels of phenobarbital. Upon discontinuation of CO2, plasma pH returned to normal and phenobarbital levels moved upward. Perhaps the most important example of an altered drug effect occurring in response to electrolyte imbalance involves digoxin, a drug for heart disease. The most serious toxicity of digoxin is production of potentially fatal dysrhythmias. The tendency of digoxin to disturb cardiac rhythm is related to levels of potassium: When potassium levels are depressed, the ability of digoxin to induce dysrhythmias is greatly increased. Accordingly, all patients receiving digoxin must undergo regular measurement of serum potassium to ensure that levels remain within a safe range. Digoxin toxicity and its relationship to potassium levels are discussed at length in Chapter 48. The experiment summarized in Table 8–1 demonstrates the development of metabolic tolerance in response to repeated administration of pentobarbital, a central nervous system depressant. The study employed two groups of rabbits, a control group and an experimental group. Rabbits in the experimental group were pretreated with pentobarbital for 3 days (60 mg/kg/day subQ) and then given an IV challenging dose (30 mg/kg) of the same drug. Drug effect (sleeping time) and plasma drug levels were then measured. The control rabbits received the challenging dose of pentobarbital but did not receive any pretreatment. As indicated in Table 8–1, the challenging dose of pentobarbital had less effect on the pretreated rabbits than on the control animals. Specifically, whereas the control rabbits slept an average of 67 minutes, the pretreated rabbits slept only 30 minutes—less than half the sleeping time seen in controls. TABLE 8–1 Development of Metabolic Tolerance as a Result of Repeated Pentobarbital Administration Data from Remmer H: Drugs as activators of drug enzymes. In Brodie BB, Erdos EG (eds): Metabolic Factors Controlling Duration of Drug Action (Proceedings of First International Pharmacological Meeting, Vol 6). New York: Macmillan, 1962:235. (See text for details.) Transdermal nitroglycerin provides a good example of tachyphylaxis. When nitroglycerin is administered using a transdermal patch, effects are lost in less than 24 hours (if the patch is left in place around the clock). As discussed in Chapter 51, the loss of effect results from depletion of a cofactor required for nitroglycerin to act. When nitroglycerin is administered on an intermittent schedule, rather than continuously, the cofactor can be replenished between doses and no loss of effect occurs. Until recently, the power of the placebo effect was unquestioned by most clinicians and researchers. However, evidence now suggests that responses to placebos may be much smaller than previously believed (Box 8–1). HAS THE PLACEBO LOST ITS EFFECT? In 1955, H. K. Beecher wrote his famous paper—“The Powerful Placebo”1—which was heralded as solid proof for the long-held (but largely unsubstantiated) belief that placebos can effectively relieve symptoms in many patients. This widely cited paper had gone unchallenged until 2001, when two Danish scientists—Hróbjartsson and Gøtzsche—wrote their own paper on the subject, titled “Is the Placebo Powerless?”2 From their research, the Danes concluded that, at least in the context of clinical trials, placebo treatment has little or no measurable effect. Who’s right? Let’s consider both papers and see if we can decide. • If the placebo response is based primarily on the clinician-patient relationship, then, even if there is a placebo response, it would be invisible in clinical trials. Why? Because subjects who receive placebo treatment and those who receive no treatment all share the same relationship with the clinician. • Placebo responses (assuming they exist) are based on the patient’s strong belief that he or she is getting an effective treatment. However, in clinical trials, there is always doubt—because all participants are aware that they may be getting a placebo, rather than the real deal. In the presence of significant doubt, the placebo effect may be greatly diminished. If this is true, then placebo effects would not be expected in clinical trials. • If placebo effects exist only in real practice—and not in clinical trials—then proving their existence may well be impossible. Why? Because we’d have to do a clinical trial to prove they exist—and we already know we can’t see them in clinical trials.
Individual variation in drug responses
Age
Pathophysiology
Kidney disease
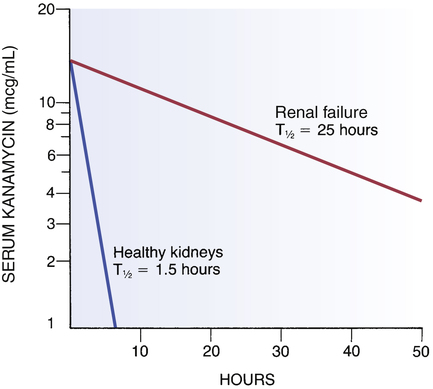
 Effect of renal failure on kanamycin half-life.
Effect of renal failure on kanamycin half-life.
Kanamycin was administered at time “0” to two patients, one with healthy kidneys and one with renal failure. Note that drug levels declined very rapidly in the patient with healthy kidneys and extremely slowly in the patient with renal failure, indicating that renal failure greatly reduced the capacity to remove this drug from the body. (T1/2 = half-life.)
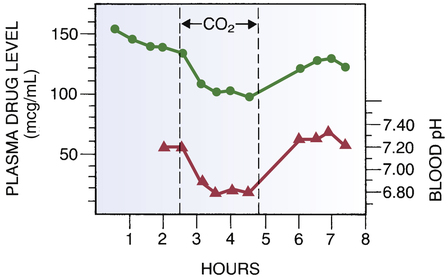
 Altered drug distribution in response to altered plasma pH.
Altered drug distribution in response to altered plasma pH.
Lower curve, Plasma (extracellular) pH. Note the decline in pH in response to inhalation of CO2. Upper curve, Plasma levels of phenobarbital. Note the decline in plasma drug levels during the period of extracellular acidosis. This decline results from the redistribution of phenobarbital into cells. (See text for details.) (Redrawn from Waddell WJ, Butler TC: The distribution and excretion of phenobarbital. J Clin Invest 36:1217, 1957.)
Tolerance
Metabolic tolerance

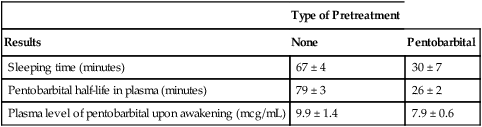
Type of Pretreatment
Results
None
Pentobarbital
Sleeping time (minutes)
67 ± 4
30 ± 7
Pentobarbital half-life in plasma (minutes)
79 ± 3
26 ± 2
Plasma level of pentobarbital upon awakening (mcg/mL)
9.9 ± 1.4
7.9 ± 0.6
Placebo effect
 BOX 8–1
BOX 8–1  SPECIAL INTEREST TOPIC
SPECIAL INTEREST TOPIC
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Individual variation in drug responses
Get Clinical Tree app for offline access
