15 Hematologic and Oncologic Emergencies Requiring Critical Care
 Be sure to check out the supplementary content available at
Be sure to check out the supplementary content available at Pearls
• Severe anemia associated with a hemoglobin (Hgb) concentration less than 5 g/dL and a hematocrit (Hct) less than 15% is likely to produce congestive heart failure (CHF). Transfuse packed red blood cells (PRBCs) slowly (3 mL/kg per hour).
• Complete bone marrow failure produces a fall in Hgb of approximately 0.7-1 g/dL per day. A more precipitous fall in Hgb is likely to be caused by bleeding or increased red blood cell (RBC) destruction.
• The risk of spontaneous bleeding is increased if the platelet count is less than 20,000/mm3 and intracranial hemorrhage is more likely once the platelet count is less than 5000/mm3.
• Rule out intracranial hemorrhage in any child with severe thrombocytopenia and unilateral headache. If hemorrhage is suspected, you will often need to begin to stabilize first; obtain scans and radiographs later.
• Acute tumor lysis syndrome produces hypocalcemia, hyperkalemia, hyperuricemia, and hyperphosphatemia. Disseminated intravascular coagulation (DIC) is also possible.
• An absolute neutrophil count less than 500/mm3 is associated with a significant risk of infection. Monitor these patients closely for evidence of infection.
• Whenever blood products are administered, label the crossmatch specimen carefully at the bedside and double-check patient and blood product identification (an extra time).
Essential anatomy and physiology
Blood Components
Blood is the fluid that sustains life. This highly specialized body fluid has many functions, including transporting oxygen and nutrition, eliminating waste, acid-base buffering, and maintaining homeostasis. The hematopoietic system consists of the bone marrow, liver, spleen, lymph nodes, and thymus gland.27 Each of these components plays a specific role in the regulation of blood.
Blood is composed of a liquid phase called plasma and a formed or cellular phase. Approximately 90% of plasma consists of water, and the remaining portions are solutes including factors that form clots (e.g., clotting factors, fibrinogen, globulins) and serum that contains electrolytes, hormones, antibodies, nutrients, and other factors. The cellular portion of the blood consists of the formed elements: RBCs, white blood cells (WBCs), and platelets (for an illustration, refer to Evolve Fig. 15-1 in the Chapter 15 Supplement on the Evolve Website).
The body’s first hematopoietic stem cell is produced in the yolk sac of the embryo. The fetal liver becomes the site for hematopoiesis at approximately the second month of fetal life. The liver is the main organ producing blood cells from the second to the fifth month of fetal life.27 At approximately the fourth month, the bone marrow begins producing blood cells and remains the production site throughout life. Bone marrow, one of the largest organs in the body, is located in the cavities of all bones.
Hematopoiesis consists of the production, differentiation, and development of blood cells; it normally occurs in the bone marrow.27 RBCs, WBCs, and platelets are all thought to arise from multipotential stem cells that inhabit the bone marrow. These stem cells have the ability to differentiate into any of the three cell lines, based on the needs of the body. The process of cell differentiation by the pluripotent stem cell is normally (i.e., in the absence of disease) self regulatory. In children, all bones produce blood cells. When bone growth ceases, often by 18 years old, only the ribs, sternum, vertebrae, and pelvis continue to produce blood cells.27
WBCs, also called leukocytes, defend the body against infectious agents. Their main function is to fight infection by migrating to the site of inflammation to assist in defending the body against foreign antigens. There are several different types of WBCs, each with a specific purpose. The differential of a complete blood cell count (CBC) reveals the percentages of neutrophils, lymphocytes, monocytes, and eosinophils. The numeric value of the WBCs is not as important as an evaluation of the absolute neutrophil count (ANC), a more accurate measure of the body’s ability to fight infection (see Neutrophils below and formula for ANC calculation in Box 15-1).
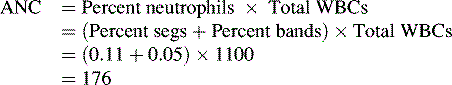
Neutrophils provide the primary defense against bacterial infections, because they engulf and destroy invading organisms. The ANC is determined from a WBC differential by multiplying the percent of neutrophils plus bands by the total WBC count (Box 15-1 shows an example of ANC calculation). In general, children with an ANC of less than 500/mm3 have a high risk of developing life-threatening bacterial infections. The risk of infection is particularly high in patients with neutropenia as the result of failure of neutrophil production, rather than those with immune-mediated neutropenia.
Clotting Cascade
Normal hemostasis is maintained by a complex balance of procoagulant and anticoagulant factors. Together, these factors provide rapid and localized control of bleeding at sites of injury while preventing the clotting process in unaffected tissues. A simplified scheme of the coagulation process is shown in Evolve Fig. 15-2 in the Chapter 15 Supplement on the Evolve Website.
Common clinical conditions
Acute Anemia
Etiology
Anemia is present when the number of circulating RBCs is reduced, resulting in decreased oxygen-carrying capacity of the blood. Anemia is the most common hematologic condition of infancy and childhood. Most patients with anemia have an underlying disease, so anemia is a symptom rather than a disease. Causes of anemia include acute or chronic blood loss, decreased production of RBCs, splenic sequestration, and hemolysis (Box 15-2).
Box 15-2 Etiology of Severe Anemia
Decreased Production
• Bone marrow replacement (leukemia, other malignancies)
• Bone marrow failure (aplastic anemia)
• Bone marrow suppression (chemotherapy, radiation)
• Transient erythroblastopenia of childhood
• Congenital red cell aplasia (Diamond-Blackfan syndrome)
• Aplastic crisis (sickle cell anemia, hemoglobin sickle cell disease, hereditary spherocytosis)
Pathophysiology
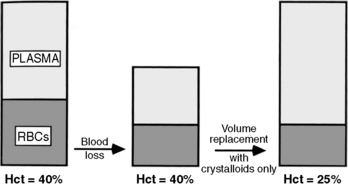
Blood Loss
When a patient experiences acute blood loss, the Hgb and Hct are not initially affected because both plasma and RBCs have been lost. If only crystalloid and colloids are used (without blood) for fluid resuscitation, then the Hct will fall because whole blood loss has been replaced with fluid that does not contain RBCs (Fig. 15-1). The patient may remain anemic until blood is administered.
Decreased RBC Production
When complete bone marrow failure is present, the Hgb will fall approximately 0.7 to 1 g/dL per day7; this usually produces a chronic rather than an acute anemia. If the Hgb or Hct falls precipitously (greater than 1 g/dL per day) in the patient with bone marrow suppression, then hemorrhage or increased RBC destruction is probably present.
Thrombocytopenia
Etiology and Pathophysiology
Patients with thrombocytopenia have a decrease in circulating platelets. This condition can occur as a result of decreased production of platelets, increased destruction of platelets, or sequestration of platelets in the spleen or liver (Box 15-3). The cause of the thrombocytopenia affects the treatment.
Box 15-3 Most Common Causes of Thrombocytopenia
Decreased Production
• Bone marrow failure (aplastic anemia)
• Bone marrow replacement (leukemia, other malignancies)
• Bone marrow suppression (chemicals, insecticides, chemotherapy, radiation therapy, chloramphenicol, alcohol)
• Hereditary thrombocytopenia (Alport’s syndrome, Fanconi syndrome, Wiskott-Aldrich syndrome)
Increased Destruction
From Turgeon ML: Clinical hematology: theory and procedures, ed 4. Philadelphia, 2005, Lippincott Williams and Wilkins.
Clinical Signs and Symptoms
Reticulated platelets (RPs) are newly synthesized platelets with increased ribonucleic acid content. These new platelets can be identified and quantified, and the RP value can aid in determining the cause of the thrombocytopenia. A decrease in RP value in the patient with thrombocytopenia suggests bone marrow suppression (i.e., platelets are not being made despite a fall in platelet count), whereas a high RP value in a patient with thrombocytopenia suggests immune-mediated thrombocytopenia (i.e., platelets are being made but are not correcting the thrombocytopenia).21
Management
Prophylactic platelet transfusion is provided if the platelet count is less than 10,000/mm3, to reduce the risk of spontaneous intracranial hemorrhage. Although published data indicate no difference in outcomes for patients with leukemia when platelet transfusion thresholds were 10,000/mm3 or 20,000/mm3 in the absence of active bleeding, institutional practices vary widely based on local practice guidelines.28 In general, lower thresholds are used for platelet transfusion for children with conditions such as fever, central nervous system tumors, bleeding, or coagulopathies or those requiring procedures.2 Patients can safely have “major invasive procedures” such as bone marrow aspirate and biopsies with platelet counts of 50,000/mm3; the thresholds can vary slightly among institutions.23,24,28
Patients with immune-mediated platelet destruction can be treated with high-dose steroids (prednisone, 4 to 8 mg/kg per day), intravenous immune gamma globulin (0.5 to 1 g/kg of immunoglobulin G), or both. Children with exceptionally low platelet counts and acute bleeding require inpatient hospitalization with close observation. Bone marrow aspirate is recommended before initiating high-dose methylprednisolone (30 to 50 mg/kg per day) to treat severe bleeding. These children may also receive a 2- or 3-day course of intravenous immune gamma globulin.13 Frequent monitoring of laboratory tests may be necessary until the platelet count improves. Monitor the patient closely for signs of bleeding.
Disseminated Intravascular Coagulation
Etiology
Disseminated intravascular coagulation is characterized by the intravascular consumption of platelets and plasma clotting factors.13 DIC is not a primary disease but a complication of other disease processes.27 DIC can complicate sepsis, hypoxemia, major trauma with severe tissue injury, malignancy, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome (HUS), extensive burns, and severe viral infections (Box 15-4).
Pathophysiology and Clinical Signs and Symptoms
DIC is characterized by an abnormal coagulation process—the entire clotting mechanism is triggered inappropriately. Unrestrained clotting causes systemic or local formation of fibrin clots. This development of fibrin clots leads to microthrombi and excessive bleeding secondary to the consumption of the platelets and clotting factors. The fibrin clot development triggers the clotting cascade process to break down the clots and fibrin that have been formed. Fibrinolysis results in the development of fibrin degradation products, which in turn will act as anticoagulants and promote more bleeding. Organ injury may be the end result of intravascular clots, which cause microvascular occlusions and anoxia.20 In addition, when the RBCs flow through obstructed vessels the end result is additional hemolysis that can lead to hemolytic anemia (Fig. 15-2).
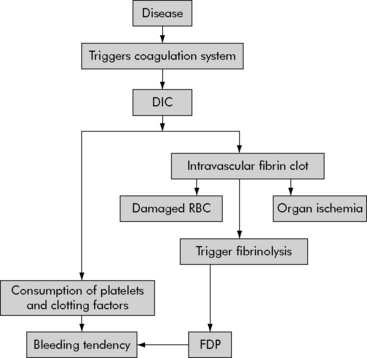
Fig. 15-2 Pathophysiology of disseminated intravascular coagulation.
DIC, Disseminated intravascular coagulation; FDP, fibrin degradation products.
(From Pagana K, Pagana T: Mosby’s manual of diagnostic and laboratory tests, St Louis, 2006, Mosby-Elsevier.)
No single test can be used to diagnose DIC. Laboratory abnormalities consistent with the diagnosis of DIC include thrombocytopenia, prolonged PT or activated partial thromboplastin time (aPTT), decreased fibrinogen, elevated fibrin degradation products (fibrin split products), and anemia (Table 15-1). The presence of fibrin monomer indicates that fibrinogen is being broken down by thrombin and that clot is being formed. A rise in fibrin monomer is an early indication of DIC.
Table 15-1 Disseminated Intravascular Coagulation Blood Tests
| Test | Result |
| Bleeding time | Prolonged |
| Platelet count | Decreased |
| Prothrombin time | Prolonged |
| Activated partial thromboplastin time | Prolonged |
| Coagulation factors | I, II, V, VIII, X, and XIII decreased |
| Fibrin degradation products | Increased |
| Red blood smear | Damaged red blood cells |
| Euglobulin lysis time | Normal or prolonged |
| D-dimer | Increased |
| Thrombin time | Prolonged |
| Fibrinopeptide A | Increased |
| Prothrombin fragment | Increased |
Hyperleukocytosis
Clinical Signs and Symptoms
Patients with hyperleukocytosis often have clinical complications from clumping of the leukemia cells in the lungs and brain,27 with resultant pulmonary sequestration or cerebral thromboembolic events. Signs and symptoms of pulmonary sequestration include hypoxia, restlessness, agitation, shortness of breath, tachypnea, cyanosis, decreased lung sounds, and increased work of breathing. Respiratory distress will persist despite oxygen administration.
Management
Perform frequent pulmonary and neurologic assessment to detect early respiratory or neurologic compromise. Evaluate oxygenation and respiratory effort, and evaluate the patient’s ability to follow commands, pupil size, and response to light. Next, immediately investigate any change in level of consciousness or headache (see Increased Intracranial Pressure in Chapter 11). In addition, monitor systemic perfusion and fluid and electrolyte balance.
Acute Tumor Lysis Syndrome
Etiology
Acute tumor lysis syndrome (ATLS) is a metabolic condition that can occur as a result of rapidly proliferating malignancies, such as Burkitt’s lymphoma, or following induction chemotherapy for malignancies that have a large tumor burden, leukemia with high WBC count, and tumors that respond rapidly to chemotherapy. ATLS can occur immediately after therapy, but most often will be observed 2 to 3 days after initiating treatment. The syndrome typically lasts approximately 7 days.4
Pathophysiology
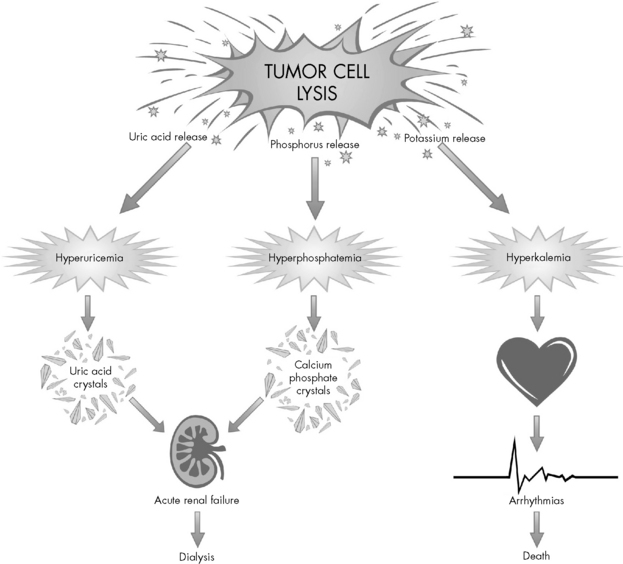
Destroyed cells release a large amount of potassium, producing hyperkalemia that can rapidly worsen with the development of renal failure (Fig. 15-3). Hyperkalemia is one of the most dangerous electrolyte imbalances, and fatal arrhythmias may occur with potassium levels greater than 7.0 mEq/L. Hyperphosphatemia can cause the formation of calcium phosphate crystals. Similar to uric acid crystals, calcium phosphate crystals can also obstruct the renal tubules compromising renal function. The precipitation of calcium phosphate crystals also may cause a secondary hypocalcemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


