4 Pharmacokinetics and Pharmacodynamics
 Be sure to check out the supplementary content available at
Be sure to check out the supplementary content available at Pearls
• Pharmacokinetics describes how the body alters drug concentration, including how the drug is dispersed in and removed from the body; it is the effect that the body has on the drug. Pharmacokinetics involves drug absorption, distribution, and elimination.
• Pharmacodynamics describes the relationship between drug concentration and drug effect; it is the effect the drug has on the body.
• The steady state is a state of equilibrium between how much of the drug is administered and how much is being removed from the body. In drugs with first-order kinetics (i.e., the systems that eliminate the drug are not saturated), steady state can be predicted from the drug half-life (t1/2).
• The drug half-life is the time it takes for half of the drug to be eliminated from the body. In each half-life, the drug concentration in the blood will fall by half. You can use the half-life to predict the time to steady state and to know when to anticipate drug effects or changes in effects.
• In general, when administering a drug by continuous infusion, a blood concentration of 90% of steady-state value is achieved in approximately four to five half-lives of the drug. Drugs with long half-lives will require a longer time to achieve steady state unless a loading dose is provided.
Principles of pharmacokinetics
The effects of drug administration vary with both the drug and the patient. There have been many attempts to model these processes using mathematical equations to guide clinical therapy. In addition, understanding of developmental changes in drug metabolism and excretion and emerging information about pharmacogenetics enable more accurate prediction of pediatric drug dosing and effects.2–5
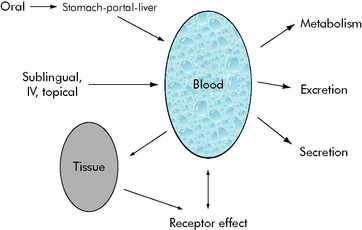
Mathematical modeling of drug paths
Michaelis-Menten Kinetics
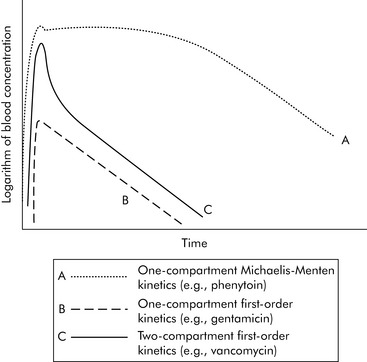
A common drug governed by Michaelis-Menten kinetics is phenytoin. With even a single dose of phenytoin, the enzymes that metabolize the drug (the cytochrome P450 enzymes) are typically saturated, so the phenytoin blood concentration will initially fall slowly after administration. However, once the blood concentration falls sufficiently, the enzymes responsible for metabolism are no longer saturated and the blood concentration will then fall quickly (Fig. 4-2, curve A). Giving too much of a drug initially or giving additional doses too soon can increase the drug concentration and risk of toxicity and prolong effects and elimination time. Implications of phenytoin kinetics with repeated dosing are discussed in the next section.
First Order Kinetics
With first-order kinetics, drugs behave similarly to radioactive decay, and elimination is described in terms of the drug’s half-life (t1/2). The drug half-life is the time it takes for half of the drug to be eliminated from the body. When a half-life is listed in a drug database, the drug has first-order kinetics (see section, Half-life). See Box 4-1 for a metaphor to further explain first- and zero-order kinetics. Many of the principles described in the following sections (e.g., time to study state, volume of distribution, filtration rates) apply chiefly to drugs with first-order kinetics.
Volume of Distribution
Generally speaking, when injecting an intravenous drug, the first compartment it occupies is the blood. If the drug is primarily distributed in the blood (e.g., gentamicin), that is where the drug remains until it is eliminated; these kinetics are described as one-compartment kinetics. The graph of the logarithm of blood concentration over time shows a rise when the drug is administered and a straight line as the drug is eliminated (see Fig. 4-2, curve B).
If a drug is distributed in the blood and the tissues (e.g., vancomycin), intravenous administration temporarily increases the concentration of the drug in the blood. Initially, blood levels decline rapidly as the drug moves into tissue, and then a more gradual decline occurs as the drug is eliminated. This drug activity is called two-compartment kinetics (see Fig. 4-2, curve C).
Implications of Multicompartment Distribution
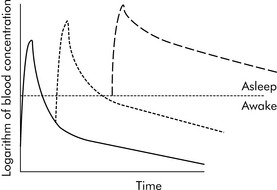
Drugs can disappear rapidly from the blood if they are distributed in the tissue. The best example of this is sodium thiopental (Pentothal). In clinical practice, a single dose of intravenous thiopental has a short effect. However, the drug has a long final half-life. The explanation for this apparent contradiction is that most thiopental elimination occurs after the drug concentration is below the level needed to keep the patient asleep (i.e., anesthetized). If several doses of thiopental are administered in a short period of time in an attempt to produce anesthesia, the tissues will become saturated and no distribution will occur. If the drug concentration increases to sufficiently high levels, the clinical effect (i.e., anesthesia) may last a long time (see Fig. 4-3).