On completion of this chapter, the reader will be able to: • Differentiate between the disorders caused by hypopituitary and hyperpituitary dysfunction. • Describe the manifestations of thyroid hypofunction and hyperfunction and the management of children with the disorders. • Distinguish between the manifestations of adrenal hypofunction and hyperfunction. • Differentiate among the various categories of diabetes mellitus. • Discuss the management and nursing care of the child with diabetes mellitus in the acute care setting. • Distinguish between a hypoglycemic and a hyperglycemic reaction. • Formulate a teaching plan for instructing the parents of a child with diabetes mellitus. The endocrine system consists of three components: (1) the cells, which send chemical messages by means of hormones; (2) the target cells, or end organs, which receive the chemical messages; and (3) the environment through which the chemicals are transported (blood, lymph, extracellular fluids) from the sites of synthesis to the sites of cellular action. The endocrine system controls or regulates metabolic processes governing energy production, growth, fluid and electrolyte balance, response to stress, and sexual reproduction (Baxter and Ribeiro, 2004). The pathophysiology review in Fig. 46-1 provides a summary of the principal pituitary hormones and their target organs. A hormone is a complex chemical substance produced and secreted into body fluids by a cell or group of cells that exerts a physiologic controlling effect on other cells (Kliegman, Stanton, St. Geme, et al., 2011). These effects may be local or distant and may affect either most cells of the body or specific “target” tissues. Hormones are released by the endocrine glands into the bloodstream, and production is regulated by a feedback mechanism. The master gland of the endocrine system is the anterior pituitary, which is in turn controlled by the hypothalamus. Some hormones, such as insulin, are regulated by other mechanisms. The pituitary gland is divided into two lobes—the anterior and the posterior lobe. Each lobe is responsible for different hormones. Disorders of the anterior pituitary hormones may be attributable to organic defects or have an idiopathic etiology and may occur as a single hormonal problem or in combination with other hormonal disorders. The clinical manifestations depend on the hormones involved and the age of onset. Panhypopituitarism is often defined clinically as the loss of all anterior pituitary hormones, leaving only posterior function intact (Toogood and Stewart, 2008). Children with panhypopituitarism should wear medical identification, such as a bracelet. Hypopituitarism is diminished or deficient secretion of pituitary hormones. The consequences of the condition depend on the degree of dysfunction and can lead to gonadotropin deficiency with absence or regression of secondary sex characteristics; growth hormone (GH) deficiency, in which children display slowed somatic growth; thyroid-stimulating hormone (TSH) deficiency, which produces hypothyroidism; and corticotropin deficiency, which results in manifestations of adrenal hypofunction. Hypopituitarism can result from any of the conditions listed in Box 46-1. The most common organic cause of pituitary undersecretion is tumors in the pituitary or hypothalamic region, especially the craniopharyngiomas. Congenital hypopituitarism can be seen in newborn infants, often as a result of birth trauma. Symptoms of hypoglycemia and seizure activity often manifest within the first 24 hours after birth (Toogood and Stewart, 2008). Idiopathic hypopituitarism, or idiopathic pituitary growth failure, is usually related to GH deficiency, which inhibits somatic growth in all cells of the body (Miller and Zimmerman, 2004). Growth failure is defined as an absolute height of less than −2 standard deviation (SD) for age or a linear growth velocity consistently less than −1 SD for age. When this occurs without the presence of hypothyroidism, systemic disease, or malnutrition, an abnormality of the GH–insulin-like growth factor (IGF-I) axis should be considered (Richmond and Rogol, 2008). Not all children with short stature have GH deficiency. In most instances, the cause is either familial short stature or constitutional growth delay. Familial short stature refers to otherwise healthy children who have ancestors with adult height in the lower percentiles. Constitutional growth delay refers to individuals (usually boys) with delayed linear growth, generally beginning as a toddler, and skeletal and sexual maturation that is behind that of age mates (Halac and Zimmerman, 2004; Miller and Zimmerman, 2004). Typically, these children will reach normal adult height. Often there is a history of a similar pattern of growth in one of the child’s parents or other family members. The untreated child will proceed through normal changes as expected on the basis of bone age. Although treatment with GH is not usually indicated, its use has become controversial, especially in relation to parental and child requests for treatment to accelerate growth. Children with hypopituitarism generally grow normally during the first year and then follow a slowed growth curve that is below the third percentile. Skeletal proportions and weight are normal for the age, but these children may appear younger than their chronologic age. Dentition is delayed, and teeth may be overcrowded and malpositioned because of the undeveloped jaw. Sexual development is usually delayed but is otherwise normal unless the gonadotropin hormones are deficient. Growth may extend into the third or fourth decade of life, but permanent height is usually diminished if the disorder is left untreated. Symptoms such as headache and vision changes may indicate the presence of a tumor. Clinical manifestations of panhypopituitarism are listed in Box 46-1. A complete diagnostic evaluation should include a family history, a history of the child’s growth patterns and previous health status, physical examination, psychosocial evaluation, radiographic surveys, and endocrine studies. Accurate measurement of height (using a calibrated stadiometer) and weight and comparison with standard growth charts are essential. Multiple height measures reflect a more accurate assessment of abnormal growth patterns (Box 46-2) (Hall, 2000). Parental height and familial patterns of growth are important clues to diagnosis. Box 46-2 Evaluating the Growth Curve Ensure reliability of measurements. Accurately obtain and plot height and weight measurements. Determine absolute height. The child’s absolute height bears some relationship to the likelihood of a pathologic condition. However, the majority of children who have a height below the lowest percentile (either the third or fifth percentile on the height curve) do not have a pathologic growth problem. Assess height velocity. The most important aspect of a growth evaluation is the observation of a child’s height over time, or height velocity. Accurate determination of height velocity requires at least 4 and preferably 6 months of observation. A substantial deceleration in height velocity (crossing several percentiles) between 3 and 12 or 13 years of age indicates a pathologic condition until proven otherwise. Determine weight-to-height relationship. Determination of the weight-to-height ratio has some diagnostic value in ascertaining the cause of growth delay in a short child. Project target height. The height of a child can be judged inappropriately short only in the context of his or her genetic potential. Determine the target height of the child with the formula: Most children achieve an adult stature within approximately 10 cm (4 inches) of the target height. Adapted from Vogiatzi MG, Copeland KC: The short child, Pediatr Rev 19(3):92–99, 1998. A skeletal survey in children younger than 3 years and radiographic examination of the hand-wrist for centers of ossification (bone age) (Box 46-3) in older children are important in evaluating growth. Definitive diagnosis is based on absent or subnormal reserves of pituitary GH. Because GH levels are variable in children, GH stimulation testing is usually required for diagnosis. Initial assessment of the serum IGF-I and IGF binding protein 3 (IGFBP3) indicates a need for further evaluation of GH dysfunction if levels are less than −1 SD below the mean for age. It is recommended that GH stimulation tests be reserved for children with low serum IGF-I and IGFBP3 levels and poor growth who do not have other causes for short stature (Richmond and Rogol, 2008). GH stimulation testing involves the use of pharmacologic agents such as levodopa, clonidine, arginine, insulin, propranolol, or glucagon to provoke the release of GH (Kliegman, Stanton, St. Geme, et al., 2011). Children with poor linear growth, delayed bone age, and abnormal GH stimulation tests are considered GH deficient. Treatment of GH deficiency caused by organic lesions is directed toward correction of the underlying disease process (e.g., surgical removal or irradiation of a tumor). The definitive treatment of GH deficiency is replacement of GH, which is successful in 80% of affected children. Biosynthetic GH is administered subcutaneously on a daily basis. Growth velocity increases in the first year and then declines in subsequent years. Final height is likely to remain less than normal (Bryant, Baxter, Cave, et al., 2007), and early diagnosis and intervention are essential (Leschek, Rose, Yanovski, et al., 2004). The decision to stop GH therapy is made jointly by the child, family, and health care team. Growth rates of less than 1 inch per year and a bone age of more than 14 years in girls and more than 16 years in boys are often used as criteria to stop GH therapy (Kliegman, Stanton, St. Geme, et al., 2011). Children with other hormone deficiencies require replacement therapy to correct the specific disorders. Children undergoing hormone replacement require additional support. The nurse should provide education for patient self-management during the school-age years. Nursing functions include family education concerning medication preparation and storage, injection sites, injection technique, and syringe disposal (see Chapter 39). Administration of GH is facilitated by family routines that include a specific time of day for the injection. Younger children may enjoy using a calendar and colorful stickers to designate received injections. Professionals and families can find resources for research, education, support, and advocacy from the Human Growth Foundation.* The treatment is expensive, but the cost is often partially covered by insurance if the child has a documented deficiency. Children with panhypopituitarism should be advised to wear medical identification at all times. If a lesion is present, surgery is performed to remove the tumor when feasible. Other therapies aimed at destroying pituitary tissue include external irradiation and radioactive implants. New pharmacologic agents have evolved and may be used in combination with other therapies (Natchtigall, Delgado, Swearingen, et al., 2008). Depending on the extent of surgical extirpation and degree of pituitary insufficiency, hormone replacement with thyroid extract, cortisone, and sex hormones may be necessary. Manifestations of sexual development before age 9 years in boys or age 8 years in girls have traditionally been considered precocious development, and these children were recommended for further evaluation (Kempers and Otten, 2002; Midyett, Moore, and Jacobson, 2003). Recent examination of the age limit for defining when puberty is precocious reveals that the onset of puberty in girls is occurring earlier than previous studies have documented (Biro, Huang, Crawford, et al., 2006; Slyper, 2006). The mean onset of puberty is 10.2 years in Caucasian girls and 9.6 years in African-American girls. Based on these findings, precocious puberty evaluation for a pathologic cause should be performed for Caucasian girls younger than 7 years or for African-American girls younger than 6 years. No change in the guidelines for evaluation of precocious puberty in boys is recommended. However, recent data suggest that boys may be beginning maturation earlier as well (Herman-Giddens, 2006; Slyper, 2006). Normally, the hypothalamic-releasing factors stimulate secretion of the gonadotropic hormones from the anterior pituitary at the time of puberty. In boys, interstitial cell–stimulating hormone stimulates Leydig cells of the testes to secrete testosterone; in girls, follicle-stimulating hormone (FSH) and luteinizing hormone (LH) stimulate the ovarian follicles to secrete estrogens (Nebesio and Eugster, 2007). This sequence of events is known as the hypothalamic-pituitary-gonadal axis. If for some reason the cycle undergoes premature activation, the child will display evidence of advanced or precocious puberty. Causes of precocious puberty are found in Box 46-4. Isosexual precocious puberty is more common among girls than boys. Approximately 80% of children with precocious puberty have central precocious puberty (CPP), in which pubertal development is activated by the hypothalamic gonadotropin-releasing hormone (GnRH) (Greiner and Kerrigan, 2006). This produces early maturation and development of the gonads with secretion of sex hormones, development of secondary sex characteristics, and sometimes production of mature sperm and ova (Lee, Houk, Ahmed, et al., 2006; Root, 2000). CPP may be the result of congenital anomalies; infectious, neoplastic, or traumatic insults to the central nervous system (CNS); or treatment of longstanding sex hormone exposure (Trivin, Couto-Silva, Sainte-Rose, et al., 2006). CPP occurs more frequently in girls and is usually idiopathic, with 95% demonstrating no causative factor (Greiner and Kerrigan, 2006; Nebesio and Eugster, 2007; Root, 2000). A CNS insult or structural abnormality is found in more than 90% of boys with CPP (Root, 2000). Peripheral precocious puberty (PPP) includes early puberty resulting from hormone stimulation other than the hypothalamic GnRH–stimulated pituitary gonadotropin release. Isolated manifestations that are usually associated with puberty may be seen as variations in normal sexual development (Greiner and Kerrigan, 2006). They appear without other signs of pubescence and are caused by excess secretion of sex hormones through the gonads or adrenal glands and may be isosexual or contrasexual. Included are premature thelarche (development of breasts in prepubertal girls), premature pubarche (premature adrenarche, early development of sexual hair), and premature menarche (isolated menses without other evidence of sexual development). Treatment of precocious puberty is directed toward the specific cause when known. In 50% of cases, precocious pubertal development regresses or stops advancing without any treatment (Carel and Leger, 2008). If needed, precocious puberty of central (hypothalamic-pituitary) origin is managed with monthly injections of a synthetic analog of luteinizing hormone–releasing hormone, which regulates pituitary secretions (Greiner and Kerrigan, 2006; Muir, 2006). The available preparation, leuprolide acetate (Lupron Depot), is given in a dosage of 0.2 to 0.3 mg/kg intramuscularly once every 4 weeks. Longer-acting formulations have recently been developed as well. Breast development regresses or does not advance, and growth returns to normal rates, enhancing predicted height. Studies suggest that not all patients attain adult targeted heights, and the addition of GH therapy may be warranted (Carel and Leger, 2008). Treatment is discontinued at a chronologically appropriate time, allowing pubertal changes to resume. Psychologic management of the patient and family is an important aspect of care. Both parents and the affected child should be taught the injection procedure. Psychologic support and guidance of the child and family are the most important aspects of management. Parents need anticipatory guidance, support and information resources, and reassurance of the benign nature of the condition (Greiner and Kerrigan, 2006; O’Sullivan and O’Sullivan, 2002). Dress and activities for the physically precocious child should be appropriate to the chronologic age. Sexual interest is not usually advanced beyond the child’s chronologic age, and parents need to understand that the child’s mental age is congruent with the chronologic age. The principal disorder of posterior pituitary hypofunction is diabetes insipidus (DI), also known as neurogenic DI, resulting from undersecretion of antidiuretic hormone (ADH), or vasopressin (Pitressin), and producing a state of uncontrolled diuresis (Makaryus and McFarlane, 2006). This disorder is not to be confused with nephrogenic DI, a rare hereditary disorder affecting primarily males and caused by unresponsiveness of the renal tubules to the hormone. Neurogenic DI may result from a number of different causes. Primary causes are familial or idiopathic; of the total cases, approximately 45% to 50% are idiopathic. Secondary causes include trauma (accidental or surgical), tumors, granulomatous disease, infections (meningitis or encephalitis), and vascular anomalies (aneurysm). Certain drugs, such as alcohol and phenytoin (diphenylhydantoin), can cause a transient polyuria. DI may be an early sign of an evolving cerebral process (De Buyst, Massa, Christophe, et al., 2007). The cardinal signs of DI are polyuria and polydipsia. In older children, signs such as excessive urination accompanied by a compensatory insatiable thirst may be so intense that the child does little more than go to the toilet and drink fluids (Cheetham and Baylis, 2002). Frequently, the first sign is enuresis. In infants, the initial symptom is irritability that is relieved with feedings of water but not milk. These infants are also prone to dehydration, electrolyte imbalance, hyperthermia, azotemia, and potential circulatory collapse. If this test result is positive, the child should be given a test dose of injected aqueous vasopressin, which should alleviate the polyuria and polydipsia. Unresponsiveness to exogenous vasopressin usually indicates nephrogenic DI. An important diagnostic consideration is to differentiate DI from other causes of polyuria and polydipsia, especially DM. DI may be the early sign of an evolving cerebral process (De Buyst, Massa, Christophe, et al., 2007). The usual treatment is hormone replacement, either with an intramuscular or subcutaneous injection of vasopressin tannate in peanut oil or with a nasal spray of aqueous lysine vasopressin (Makaryus and McFarlane, 2006; Verbalis, 2003). The injectable form has the advantage of lasting 48 to 72 hours, which affords the child a full night’s sleep. However, it has the disadvantage of requiring frequent injections and proper preparation of the drug. The disorder that results from hypersecretion of ADH from the posterior pituitary hormone is known as syndrome of inappropriate ADH secretion (SIADH). It is observed with increased frequency in a variety of conditions, especially those involving infections, tumors, or other CNS disease or trauma, and is the most common cause of hyponatremia in the pediatric population (Lin, Liu, and Lim, 2005, Rivkees, 2008). Hypothyroidism is one of the most common endocrine problems of childhood. It may be either congenital or acquired and represents a deficiency in secretion of TH (Foley, 2001). Beyond infancy, primary hypothyroidism may be caused by a number of defects. For example, a congenital hypoplastic thyroid gland may provide sufficient amounts of TH during the first year or two but be inadequate when rapid body growth increases demands on the gland. A partial or complete thyroidectomy for cancer or thyrotoxicosis can leave insufficient thyroid tissue to furnish hormones for body requirements. Radiotherapy for Hodgkin disease or other malignancies may lead to hypothyroidism (Pizzo and Poplack, 2010). Infectious processes may cause hypothyroidism. It can also occur when dietary iodine is deficient, although it is now rare in the United States because iodized salt is a readily available source of the nutrient. Clinical manifestations depend on the extent of dysfunction and the child’s age at onset. Primary congenital hypothyroidism is characterized by low levels of circulating THs and raised levels of TSH at birth (Macchia, 2000). If left untreated, congenital hypothyroidism causes decreased mental capacity. Improvements in newborn screening have led to earlier detection and prevention of complications (American Academy of Pediatrics [AAP], 2006). The GnRH test and baseline measurement of gonadotropin and sex hormone serum concentrations at 3 months of age are promising options for assessment of hypothalamic-pituitary-gonadal function in infants with congenital hypothyroidism (van Tijn, Schroor, Delemarre-van de Waal, et al., 2007). The presenting symptoms are decelerated growth from chronic deprivation of TH or thyromegaly. Impaired growth and development are less severe when hypothyroidism is acquired at a later age, and because brain growth is nearly complete by 2 to 3 years of age, intellectual disability and neurologic sequelae are not associated with juvenile hypothyroidism. Other manifestations are myxedematous skin changes (dry skin, puffiness around the eyes, sparse hair), constipation, lethargy, and mental decline (Box 46-5). Therapy is TH replacement, the same as for hypothyroidism in infants, although the prompt treatment needed in infants is not required in children. l-thyroxine is administered over a period of 4 to 8 weeks to avoid symptoms of hyperthyroidism. Researchers have found that children treated early continue to have mild delays in reading, comprehension, and arithmetic but catch up by grade six (Rovet and Ehrlich, 2000). However, adolescents may demonstrate problems with memory, attention, and visuospatial processing. A goiter is an enlargement or hypertrophy of the thyroid gland. It may occur with deficient (hypothyroid), excessive (hyperthyroid), or normal (euthyroid) TH secretion. It can be congenital or acquired. Congenital disease occurs as a result of maternal administration of antithyroid drugs or iodides during pregnancy or as an inborn error of TH production. Acquired disease can result from increased secretion of pituitary TSH in response to decreased circulating levels of TH or from infiltrative neoplastic or inflammatory processes. In most children, goiter is caused by chronic autoimmune thyroiditis (de Vries, Bulvik, and Phillip, 2009). In areas where dietary iodine (essential for TH production) is deficient, goiter can be endemic. Large goiters are identified by their obvious appearance. In older children, each lobe of the thyroid should be approximately the same as the terminal phalanx of the child’s thumb (de Vries, Bulvik, and Phillip, 2009). Smaller nodules may be evident only on palpation. Benign enlargement of the thyroid gland may occur during adolescence and should not be confused with pathologic states. Nodules rarely are caused by a cancerous tumor but always require evaluation. Questions regarding exposure to radiation should be included in the assessment. Lymphocytic thyroiditis (Hashimoto disease, chronic autoimmune thyroiditis) is the most common cause of thyroid disease in children and adolescents and accounts for the largest percentage of juvenile hypothyroidism (Szymborska and Staroszczyk, 2000). It accounts for many of the enlarged thyroid glands formerly designated thyroid hyperplasia of adolescence or adolescent goiter. Although it can occur during the first 3 years of life, it occurs more frequently after age 6 years. It reaches a peak incidence during adolescence, and there is evidence that the disease is self-limiting. The presence of a goiter and elevated thyroglobulin antibody with progressive increase in both thyroid peroxidase antibody and TSH may be predictive factors for future development of hypothyroidism (Radetti, Gottardi, Bona, et al., 2006). The presence of the enlarged thyroid gland is usually detected during a routine examination, although it may be noted by parents when the child swallows. In most children, the entire gland is enlarged symmetrically (although it may be asymmetric) and is firm, freely movable, and nontender. There may be manifestations of moderate tracheal compression (sense of fullness, hoarseness, and dysphagia), but it is extremely rare for a nontoxic diffuse goiter to enlarge to the extent that it causes mechanical obstruction. Most children are euthyroid, but some display symptoms of hypothyroidism, including delayed growth and puberty and declining school performance. Other signs suggestive of thyroiditis are found in Box 46-6. The largest percentage of hyperthyroidism in childhood is caused by Graves disease, which is usually associated with an enlarged thyroid gland and exophthalmos (Ma, Xie, Kuang, et al., 2006; Streetman and Khanderia, 2004; Thompson, 2002). Most cases of Graves disease in children occur between ages 6 and 15 years, with a peak incidence at 12 to 14 years of age, but the disease may be present at birth in children of thyrotoxic mothers. The incidence is 5 times higher in girls than in boys. The hyperthyroidism of Graves disease is apparently caused by an autoimmune response to TSH receptors, but no specific etiology has been identified. There is definitive evidence for familial association, with a high concordance incidence in twins. Patients with Graves disease possess the histocompatibility antigens A1, B8, and DR3 (Dallas and Foley, 2003; Simmonds, Howson, Heward, et al., 2005). There may be an association with other autoimmune diseases such as rheumatoid arthritis and lupus. The development of manifestations is highly variable. Signs and symptoms develop gradually, with an interval between onset and diagnosis of approximately 6 to 12 months. The principal clinical features are excessive motion, including irritability, hyperactivity, short attention span, tremors, insomnia, and emotional lability. Clinical manifestations are presented in Box 46-7. Exophthalmos (protruding eyeballs), which is observed in many children, is accompanied by a wide-eyed staring expression, increased blinking, eyelid lag, lack of convergence, and absence of wrinkling of the forehead when looking upward. As protrusion of the eyeball increases, the child may not be able to completely cover the cornea with the eyelid. Visual disturbances may include blurred vision and loss of visual acuity. Ophthalmopathy can develop long before or after the onset of hyperthyroidism. A consistent pathogenic link between them has not been identified. It is now thought that Graves ophthalmopathy is a disorder of autoimmune origin caused by a complex interplay of endogenous and environmental factors (Bartalena, Tanda, Piantanida, et al., 2003). The presence of a thyroid mass in a child requires a thorough history, including inquiry into prior irradiation to the head and neck and exposure to a goitrogen. The diagnosis is established on the basis of increased levels of T4 and T3. TSH is suppressed to unmeasurable levels (Ma, Xie, Kuang, et al., 2006). Graves disease is confirmed by measurement of thyroid-stimulating immunoglobulin. Therapy for hyperthyroidism is controversial, but all methods are directed toward slowing the rate of hormone secretion. The three acceptable modes available are antithyroid drugs (methimazole), which interferes with the biosynthesis of TH; subtotal thyroidectomy; and ablation with radioiodine (131I iodide) (Rivkees and Cornelius, 2003; Streetman and Khanderia, 2004). Each is effective, but each has advantages and disadvantages. Pharmacologic therapy may induce a remission, and treatment may be discontinued. However, relapse may occur. Radioactive iodine ablation is usually effective but response may be slower, and there have been concerns about a possible link to thyroid cancer in younger children. Surgery is often used when other treatments are not effective. These children require lifelong monitoring. The American Thyroid Association* has an extensive website with information related to prevention, treatment, and cure of thyroid disease. The initial nursing objective is identification of children with hyperthyroidism. Because the clinical manifestations often appear gradually, the goiter and ophthalmic changes may not be noticed and the excessive activity may be attributed to behavioral problems. Nurses in ambulatory settings, particularly schools, need to be alert to signs that suggest this disorder, especially weight loss despite an excellent appetite, academic difficulties resulting from a short attention span and inability to sit still, unexplained fatigue and sleeplessness, and difficulty with fine motor skills such as writing. Exophthalmos may develop long before the onset of signs and symptoms of hyperthyroidism and may be the only presenting sign (Thompson, 2002). Exophthalmos is less common in adults than in children (Jospe, 2001). • Chvostek sign—facial muscle spasm elicited by tapping the facial nerve in the region of the parotid gland • Trousseau sign—carpal spasm elicited by pressure applied to nerves of the upper arm • Tetany—Carpopedal spasm (sharp flexion of wrist and ankle joints), muscle twitching, cramps, seizures, and stridor The parathyroid glands secrete PTH, the main function of which, along with vitamin D and calcitonin, is homeostasis of serum calcium concentration (Perheentupa, 2003). The effect of PTH on calcium is opposite that of calcitonin. The net result of the integrated action of PTH and vitamin D is maintenance of serum calcium levels within a narrow normal range and the mineralization of bone. Secretion of PTH is controlled by a negative feedback system involving the serum calcium ion concentration. Low ionized calcium levels stimulate PTH secretion, causing absorption of calcium by the target tissues; high ionized calcium concentrations suppress PTH. Hypoparathyroidism is a spectrum of disorders that result in deficient PTH. Congenital hypoparathyroidism may be caused by a specific defect in the synthesis or cellular processing of PTH or by aplasia or hypoplasia of the gland (Perheentupa, 2003). Pseudohypoparathyroidism occurs when there is a genetic defect in the cellular receptors to PTH. The result is normal parathyroid gland and elevated PTH levels. Abnormal calcium and phosphorus levels are not affected by administration of PTH. These children typically have a short, stocky build; a round face; and abnormally shaped hands and fingers. Other endocrine dysfunction may be found concurrently (Shoback, 2008). Clinical signs of hypoparathyroidism are found in Box 46-8. Muscle cramps are an early symptom, progressing to numbness, stiffness, and tingling in the hands and feet. A positive Chvostek or Trousseau sign or laryngeal spasms may be present. Convulsions with loss of consciousness may occur. These episodes may be preceded by abdominal discomfort, tonic rigidity, head retraction, and cyanosis. Headaches and vomiting with increased intracranial pressure and papilledema may occur and may suggest a brain tumor (Kliegman, Stanton, St. Geme, et al., 2011). The objective of treatment is to maintain normal serum calcium and phosphate levels with minimal complications. Acute or severe tetany is corrected immediately by IV and oral administration of calcium gluconate and follow-up daily doses to achieve normal levels. Twice-daily serum calcium measurements are taken to monitor the efficacy of therapy and prevent hypercalcemia. When diagnosis is confirmed, vitamin D therapy is begun. Vitamin D therapy is somewhat difficult to regulate because the drug has a prolonged onset and a long half-life. Some authorities advocate beginning with a lower dose with stepwise increases and careful monitoring of serum calcium until stable levels are achieved. Others prefer rapid induction with higher doses and rapid reduction to lower maintenance levels (Cooper and Gittoes, 2008; Kliegman, Stanton, St. Geme, et al., 2011). Hyperparathyroidism is rare in childhood but can be primary or secondary. The most common cause of primary hyperparathyroidism is adenoma of the gland (Kliegman, Stanton, St. Geme, et al., 2011). The most common causes of secondary hyperparathyroidism are chronic renal disease, renal osteodystrophy, and congenital anomalies of the urinary tract. The common factor is hypercalcemia. The clinical signs of hyperparathyroidism are listed in Box 46-9.
Endocrine Dysfunction
The Endocrine System
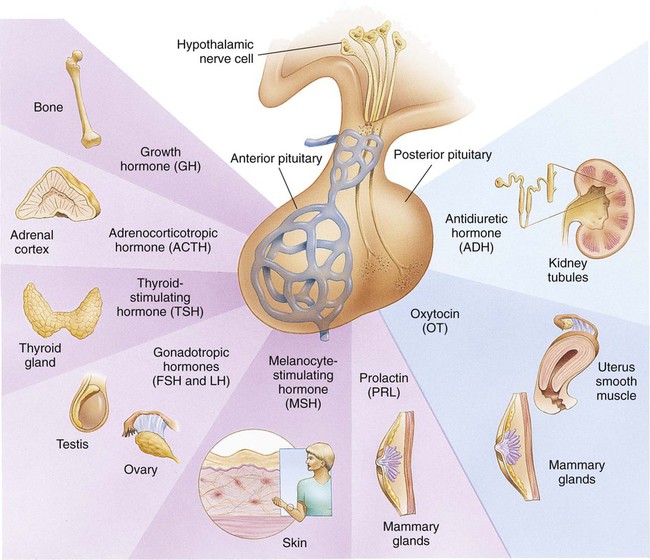
Hormones
Disorders of Pituitary Function
 Nursing Alert
Nursing Alert
Hypopituitarism
Clinical Manifestations
Diagnostic Evaluation
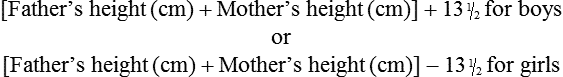
Therapeutic Management
Care Management
Child and Family Support.
Pituitary Hyperfunction
Therapeutic Management
Care Management
Precocious Puberty
Therapeutic Management
Care Management
Diabetes Insipidus
Diagnostic Evaluation
Therapeutic Management
Care Management
Syndrome of Inappropriate Antidiuretic Hormone
Disorders of Thyroid Function
Juvenile Hypothyroidism
Care Management
Goiter
Care Management
Lymphocytic Thyroiditis
Care Management
Hyperthyroidism
Diagnostic Evaluation
Therapeutic Management
Care Management
 Nursing Alert
Nursing Alert
Disorders of Parathyroid Function
Hypoparathyroidism
Therapeutic Management
Care Management
Hyperparathyroidism
 Nursing Alert
Nursing Alert Nursing Alert
Nursing Alert Nursing Alert
Nursing Alert Nursing Alert
Nursing Alert Nursing Alert
Nursing Alert
 Nursing Alert
Nursing Alert


