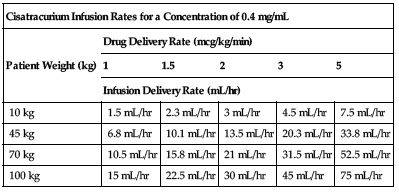
| Cisatracurium Infusion Rates for a Concentration of 0.4 mg/mL | |||||
| Patient Weight (kg) | Drug Delivery Rate (mcg/kg/min) | ||||
| 1 | 1.5 | 2 | 3 | 5 | |
| Infusion Delivery Rate (mL/hr) | |||||
| 10 kg | 1.5 mL/hr | 2.3 mL/hr | 3 mL/hr | 4.5 mL/hr | 7.5 mL/hr |
| 45 kg | 6.8 mL/hr | 10.1 mL/hr | 13.5 mL/hr | 20.3 mL/hr | 33.8 mL/hr |
| 70 kg | 10.5 mL/hr | 15.8 mL/hr | 21 mL/hr | 31.5 mL/hr | 52.5 mL/hr |
| 100 kg | 15 mL/hr | 22.5 mL/hr | 30 mL/hr | 45 mL/hr | 75 mL/hr |
Actions
A nondepolarizing skeletal muscle relaxant with intermediate onset and duration of action. An isomer of atracurium (Tracrium) with three times its potency at a mg-for-mg dose. In contrast to most of the other neuromuscular blocking agents, cisatracurium has no clinically significant effect on HR or BP with usual doses even in patients with serious cardiovascular disease, and it also does not produce a dose-related histamine release. Causes paralysis by interfering with neural transmission at the myoneural junction. Produces maximum neuromuscular blockade within 1.5 to 3 minutes and lasts about 50 minutes in adults. Recovery to 75% usually occurs within 30 minutes. Metabolized by a process that mostly bypasses both the kidney and the liver. Forms specific metabolites (e.g., alcohol, laudanosine) that do not have neuromuscular blocking activity. Because of reduced dose requirements, laudanosine accumulation is lower than with atracurium, lowering the potential of seizures. Eliminated renally, primarily as metabolites.
Indications and uses
Adjunctive to general anesthesia for inpatients and outpatients to facilitate endotracheal intubation and to relax skeletal muscles during surgery. ■ Relax skeletal muscles during mechanical ventilation in ICU.
Contraindications
Known hypersensitivity to cisatracurium, other bis-benzylisoquinolinium compounds (e.g., atracurium), and benzyl alcohol (some preparations contain benzyl alcohol).
Precautions
For IV use only. ■ Administered by or under the observation of the anesthesiologist. Adequate facilities, emergency resuscitation drugs and equipment, neuromuscular blocking antagonists (e.g., anticholinesterase agents [e.g., neostigmine, edrophonium]), and atropine must always be available. ■ Not recommended for rapid sequence intubation; succinylcholine is usually the drug of choice. ■ Severe anaphylactic reactions have been reported with neuromuscular blocking agents; some have been fatal. Use caution in patients who have had an anaphylactic reaction to another neuromuscular blocking agent (depolarizing or nondepolarizing); cross-reactivity has occurred. ■ Myasthenia gravis and other neuromuscular diseases increase sensitivity to cisatracurium. Can cause critical reactions. ■ Sensitivity may be decreased in patients with burns or paralysis. See Dose Adjustments and Monitor. ■ In patients with renal or hepatic disease, half-life of metabolites is longer, and concentrations may be higher with long-term administration. ■ Did not trigger malignant hypertension (MH) in susceptible pigs at doses above those required for humans, but has not been studied in MH-susceptible humans. ■ Respiratory depression with propofol (Diprivan) or morphine may be preferred in some patients requiring mechanical ventilation. ■ Will not counteract the bradycardia produced by many anesthetic agents or vagal stimulation. ■ Transient hypotension and CNS excitation (generalized muscle twitching to seizures) have been reported rarely in ICU patients undergoing prolonged therapy.
Monitor:
This drug produces apnea. Controlled artificial ventilation with oxygen must be continuous and under direct observation at all times. Maintain a patent airway. ■ Use a peripheral nerve stimulator to monitor drug effect, determine the need for additional doses, confirm recovery from neuromuscular block, and avoid overdose. Place on a nonparalyzed limb in patients with paralysis. ■ Monitor vital signs and ECG continuously. ■ Has no analgesic properties or effect on consciousness. Use in conjunction with anesthesia, sedation, or analgesia as indicated. ■ Action potentiated by hypokalemia and some carcinomas. ■ Action may be potentiated or antagonized by dehydration, electrolyte imbalance, body temperature, or acid-base imbalance.
Maternal/child:
Category B: use in pregnancy only if clearly needed. Safety for use during labor and delivery not established. ■ Use caution during breast-feeding; probably best to defer breast-feeding until after full recovery. ■ Safety for use in infants under 1 month of age not established. ■ 10 mL (2 mg/mL) multiple-dose vials contain benzyl alcohol; do not use in newborns. ■ In pediatric patients 2 to 12 years of age, onset is faster, duration shorter, and recovery faster than in adults. In infants 1 month to 23 months of age, onset is faster; however, duration and recovery are similar to adults. ■ For induction of anesthesia in pediatric patients 1 month to 12 years of age, intubation was facilitated more reliably when cisatracurium was used in combination with halothane anesthesia than when used in combination with opioids and nitrous oxide. ■ Rare incidences of wheezing, laryngospasm, bronchospasm, rash, and itching have been reported in pediatric patients.
Elderly:
Safely administered even in patients with significant cardiac disease. ■ Response similar to that seen in younger adults; however, greater sensitivity of some older individuals cannot be ruled out. ■ Onset to complete neuromuscular block slightly slower; delay intubation until fully effective. Recovery may be slower.
Drug/lab interactions
Potentiated by general anesthetics (e.g., enflurane, isoflurane), many antibiotics (e.g., aminoglycosides [kanamycin, gentamicin], lincosamides [clindamycin (Cleocin)], polypeptides [bacitracin, colistimethate], tetracyclines), muscle relaxants, diuretics, lithium, local anesthetics, magnesium sulfate, procainamide (Pronestyl), quinidine, succinylcholine, and others. May need to reduce initial or maintenance dose of cisatracurium; use with caution. ■ Antagonized by acetylcholine and anticholinesterases. ■ Duration of neuromuscular block may be shorter and dose requirements may be higher during maintenance infusion in patients stabilized on carbamazepine (Tegretol) or phenytoin (Dilantin). ■ Time to onset of maximum block is faster when succinylcholine is given before cisatracurium. Succinylcholine must show signs of wearing off before cisatracurium is given. Use caution.
Side effects
Bradycardia, bronchospasm, flushing, hypotension, and rash occurred in fewer than 1% of patients. Both inadequate and/or prolonged neuromuscular blocks have been reported. Rare reports of seizures with similar agents in ICU could be caused by accumulated laudanosine, other conditions, or medications. Excessive dosing or prolonged action may result in respiratory insufficiency or apnea. Airway closure may be caused by relaxation of epiglottis, pharynx, and tongue muscles. Hypersensitivity reactions have been reported.
Antidote
Side effects can be medical emergencies. Treat symptomatically. Maintain a patent airway and continuous controlled artificial ventilation and oxygenation until full recovery is ensured. The more profound the neuromuscular block, the longer it will take until recovery begins. Recovery from neuromuscular block must be confirmed by a peripheral nerve stimulator before anticholinesterase agents (e.g., neostigmine or edrophonium [Enlon]) can be given with an anticholinergic agent (e.g., atropine) to reverse the muscle relaxation. Neostigmine 0.04 to 0.07 mg/kg at 10% recovery in conjunction with atropine should be effective in 9 to 10 minutes. Edrophonium 1 mg/kg at 25% recovery in conjunction with atropine (Enlon-Plus is edrophonium and atropine combined) should be effective in 3 to 5 minutes. Confirm recovery by 5-second head lift and grip strength. Recovery may be inhibited by cachexia, carcinomatosis, debilitation, or the concomitant use of certain drugs; see Drug/Lab Interactions. Resuscitate as necessary.
Cisplatin 
(sis-PLAH-tin)
CDDP
Antineoplastic (alkylating agent)
pH 3.5 to 6
Usual dose
Prehydration required; see Precautions and Monitor. See Dose Adjustments. May be given in combination with amifostine (Ethyol) to reduce nephrotoxicity and neurotoxicity of cisplatin. See amifostine monograph. Administration as a 6- to 8-hour infusion with intravenous hydration and mannitol has been used to reduce nephrotoxicity. Doses greater than 100 mg/M2 once every 3 to 4 weeks are rarely used.
Metastatic testicular tumors:
Used in combination with other approved chemotherapeutic agents. 20 mg/M2 daily for 5 days per cycle.
Metastatic ovarian tumors:
75 to 100 mg/M2 on Day 1 every 4 weeks. Used in combination with cyclophosphamide (Cytoxan) 600 mg/M2 IV on Day 1 every 4 weeks. Another regimen (unlabeled in cisplatin prescribing information) uses paclitaxel 135 mg/M2 (as a 24-hour infusion) followed by cisplatin 75 mg/M2. Both agents are given once every 3 weeks for 6 courses. See paclitaxel monograph; premedication required. Used as a single agent, the dose of cisplatin is 100 mg/M2 every 4 weeks.
First-line treatment of ovarian cancer (unlabeled in cisplatin prescribing information):
Given in combination with paclitaxel as follows: give paclitaxel (Taxol) 135 mg/M2 as an infusion over 24 hours. Follow with cisplatin 75 mg/M2 as an infusion over 6 to 8 hours. Repeat every 3 weeks. See paclitaxel monograph; premedication required. Other dose combinations and infusion times are being used.
Advanced bladder cancer:
50 to 70 mg/M2 once every 3 to 4 weeks. 50 mg/M2 is recommended once every 4 weeks for patients heavily pretreated with radiation or chemotherapy. Numerous other doses and combinations are used.
Non–small-cell lung cancer (unlabeled in cisplatin prescribing information):
Given in combination with gemcitabine as follows: gemcitabine (Gemzar) 1,000 mg/M2 as an infusion on Days 1, 8, and 15 of each 28-day cycle. Follow the gemcitabine infusion on Day 1 with cisplatin 100 mg/M2. See gemcitabine monograph; other dosing schedules are in use. Another regimen uses a combination of paclitaxel and cisplatin as follows: paclitaxel 135 mg/M2 as an infusion over 24 hours followed by cisplatin 75 mg/M2 over 6 to 8 hours. Repeat every 3 weeks. See paclitaxel monograph; premedication required. Also used with docetaxel 75 mg/M2 infused over 1 hour followed immediately by an infusion of cisplatin 75 mg/M2 over 30 to 60 minutes. Repeat every 3 weeks. See docetaxel monograph; premedication required.
Dose adjustments
All doses adjusted based on prior radiation therapy or chemotherapy. ■ Repeat doses may not be given unless SCr is below 1.5 mg/100 mL and/or BUN is below 25 mg/100 mL. Renal toxicity becomes more prolonged and severe with repeated courses. Renal function must return to normal before next dose is given. ■ Platelets should be 100,000/mm3 and leukocytes 4,000/mm3; verify auditory acuity as within normal limits. ■ Dosing should be cautious in the elderly. Lower-end initial doses may be indicated. Consider decrease in cardiac, hepatic, and renal function; concomitant disease; or other drug therapy; see Elderly.
Dilution
Specific techniques required; see precautions.
Available in liquid form, 1 mg/mL. Withdraw desired dose. Immediately before use, manufacturer recommends diluting a single dose in 2 liters of D51/2NS or D51/3NS containing 37.5 Gm of mannitol. Do not use D5W. Will decompose if adequate chloride ion not available. Is also diluted in smaller amounts of NS (100 to 500 mL). Do not use needles or IV tubing with aluminum parts to administer; a precipitate will form, and potency will decrease. See Monitor for additional optional additives.
Storage:
Cisplatin remaining in multidose vial is stable at CRT for 28 days protected from light or 7 days under fluorescent light. Do not refrigerate. Protect from light if it will not be used within 6 hours.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Do not use needles, IV sets, or equipment containing aluminum.” Aluminum reacts with cisplatin, causing precipitate formation and loss of potency. A precipitate will form if reconstituted solutions are refrigerated.
One source suggests the following compatibilities:
Additive:
Carboplatin (Paraplatin), cyclophosphamide (Cytoxan), etoposide (VePesid), ifosfamide (Ifex), leucovorin calcium, magnesium sulfate, mannitol, ondansetron (Zofran), paclitaxel (Taxol). Another source adds carmustine (BiCNU).
Y-site:
Allopurinol (Aloprim), anidulafungin (Eraxis), aztreonam (Azactam), bleomycin (Blenoxane), caspofungin (Cancidas), cladribine (Leustatin), cyclophosphamide (Cytoxan), doripenem (Doribax), doxorubicin (Adriamycin), doxorubicin liposomal (Doxil), droperidol (Inapsine), etoposide phosphate (Etopophos), filgrastim (Neupogen), fludarabine (Fludara), fluorouracil (5-FU), furosemide (Lasix), gemcitabine (Gemzar), granisetron (Kytril), heparin, leucovorin calcium, linezolid (Zyvox), melphalan (Alkeran), methotrexate, metoclopramide (Reglan), mitomycin (Mutamycin), ondansetron (Zofran), paclitaxel (Taxol), palonosetron (Aloxi), pemetrexed (Alimta), propofol (Diprivan), sargramostim (Leukine), teniposide (Vumon), topotecan (Hycamtin), vinblastine, vincristine, vinorelbine (Navelbine).
Rate of administration
Administer as a slow IV infusion; should not be given as a rapid IV injection. Rates vary based on protocol. Manufacturer suggests administering each 1 liter of infusion solution over 3 to 4 hours. Give total dose (2 liters) over 6 to 8 hours. Rate must be sufficient to maintain hydration and diuresis. Infusion times of 30 to 120 minutes are common, but infusion time has also been extended to 24 hours/dose. One source recommends a maximum rate not to exceed 1 mg/min. Too-rapid administration increases nephrotoxicity and ototoxicity.
Actions
A heavy metal complex (platinum and chloride atoms). Has properties similar to alkylating agents and is cell-cycle nonspecific. Inhibits DNA synthesis by formation of DNA cross-links. Concentration is highest in liver, prostate, and kidney; somewhat lower in bladder, muscle, testicle, pancreas, and spleen. Heavily protein bound. Only one fourth to one half of the drug is excreted in the urine by the end of 5 days. Platinum may be present in tissues for as long as 180 days after the last administration. Secreted in breast milk.
Indications and uses
Treatment of metastatic testicular tumors; used in combination therapy with other approved chemotherapy agents in patients who have already received appropriate surgical and/or radiotherapeutic procedures. ■ Treatment of metastatic ovarian tumors; used in combination therapy with other approved chemotherapy agents in patients who have already received appropriate surgical and/or radiotherapeutic procedures. ■ Used as a single agent as secondary treatment in patients with metastatic ovarian tumors refractory to standard chemotherapy who have not previously received cisplatin therapy. ■ Used as a single agent for treatment of patients with transitional cell bladder cancer that is no longer amenable to local treatment such as surgery and/or radiotherapy. ■ Is used in specific combinations with other chemotherapeutic drugs.
Unlabeled uses:
First-line therapy for treatment of advanced cancer of the ovary in combination with paclitaxel. ■ First-line treatment of patients with inoperable, locally advanced, or metastatic non–small-cell lung cancer (NSCLC) in combination with gemcitabine, paclitaxel, or docetaxel. ■ Treatment of cancers of the brain, adrenal cortex, breast, cervix, uterus, endometrium, head and neck, esophagus, lung, and liver; osteogenic sarcomas; and numerous other malignancies.
Contraindications
Hypersensitivity to cisplatin or other platinum-containing compounds, myelosuppressed patients, pre-existing impaired renal function, or hearing deficit.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of the physician specialist. ■ Adequate facilities and emergency resuscitation equipment and supplies must always be available. ■ Renal toxicity can be cumulative and may be severe. ■ Other major dose-related toxicities include myelosuppression, nausea, and vomiting. ■ Ototoxicity (tinnitus, loss of high-frequency hearing, and/or deafness) can be significant and may be more pronounced in pediatric patients. ■ Anaphylaxis has been reported and may occur within minutes of cisplatin administration. ■ Labeling changed to read, “Doses greater than 100 mg/M2/cycle once every 3 to 4 weeks are rarely used.” This is an effort to eliminate serious errors resulting from confusion with carboplatin (Paraplatin). Flip-off seal on vial now says, “Call Dr. if dose greater than 100 mg/M2/cycle.” ■ Neuropathies may occur with higher doses, greater frequency of average doses, or prolonged therapy. Usually occur after prolonged therapy but have been reported after a single dose. If symptoms of neuropathy are observed, discontinue cisplatin. ■ See Elderly.
Monitor:
Obtain baseline CBC, SCr, BUN, CrCl and calcium, magnesium, potassium, and sodium levels. Repeat CBC weekly and other listed labs before each subsequent cycle. ■ Hydrate patient with 1 to 2 L of infusion fluid for 8 to 12 hours before cisplatin administration. Urine output should exceed 100 to 150 mL/hr. ■ Maintain adequate hydration and urine output of at least 100 to 200 mL/hr for 24 hours after each dose. ■ Nausea and vomiting are frequently severe and prolonged (up to a week). May begin within 1 to 4 hours of administration or may be delayed. Prophylactic administration of antiemetics recommended. Fosaprepitant (Emend), ondansetron (Zofran), metoclopramide (Reglan), or dexamethasone are effective in most patients. ■ Ototoxicity is cumulative; test hearing before administration and regularly during treatment. Ototoxicity increased in pediatric patients. ■ Monitor uric acid levels before and during treatment and maintain hydration. Allopurinol and alkalinization of urine may be indicated. ■ Monitor for anaphylactic-like reactions (e.g., bronchoconstriction, facial edema, hypotension, tachycardia). ■ Monitor liver function periodically. ■ Perform neurologic exams on a regular basis. Neuropathy may present as paresthesias, areflexia, and loss of proprioception and vibratory sensation. ■ Replace depleted electrolytes as necessary. ■ Observe closely for signs of infection. Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. ■ Monitor infusion site carefully during infusion; local soft tissue toxicity has been reported with extravasation. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood).
Patient education:
Nonhormonal birth control recommended. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy; can cause fetal harm. Has a mutagenic potential. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established. ■ Ototoxicity increased in pediatric patients. All pediatric patients should have audiometric monitoring performed before initiation of therapy, before each dose, and for several years after therapy.
Elderly:
Dose selection should be cautious; see Dose Adjustments. ■ Response (e.g., effectiveness) is similar to younger adults, but length of survival may be shorter. ■ Incidence of myelosuppression (e.g., severe leukopenia, neutropenia, thrombocytopenia), infectious complications, nephrotoxicity, and peripheral neuropathy may be increased.
Drug/lab interactions
Ototoxicity and nephrotoxicity are potentiated with other ototoxic or nephrotoxic agents (e.g., aminoglycosides [e.g., gentamicin] and loop diuretics [e.g., furosemide (Lasix), ethacrynic acid (Edecrin)]). Concurrent use not recommended (Lasix is used to control fluid overload; use caution). ■ Serum levels of anticonvulsant agents (e.g., phenytoin [Dilantin]) may become subtherapeutic when used concurrently with cisplatin. Monitor anticonvulsant levels; increased doses may be indicated. ■ Bone marrow toxicity increased with other antineoplastic agents and/or radiation therapy. ■ Synergistic with etoposide (VePesid); may be beneficial. ■ May affect renal excretion and increase toxicity of many drugs (e.g., bleomycin, methotrexate). ■ Response duration may be shortened with concurrent use of pyridoxine (vitamin B6) and altretamine (Hexalen). ■ Do not administer live virus vaccines to patients receiving antineoplastic agents.
Side effects
Are frequent; can occur with the initial dose and will become more severe with succeeding doses. Dose-related and cumulative renal insufficiency, including renal failure, is the major dose-limiting toxicity (often noted during the second week following a dose). Acute leukemia, alopecia, anaphylaxis (facial edema, hypotension, tachycardia, and wheezing within minutes of administration), asthenia, cardiac abnormalities, dehydration, diarrhea, electrolyte disturbances (hypocalcemia, hypokalemia, hypomagnesemia, hyponatremia, hypophosphatemia), elevated serum amylase, hemolytic anemia, hepatotoxicity, hyperuricemia, malaise, myelosuppression, nausea and vomiting (acute or delayed), syndrome of inappropriate antidiuretic hormone (SIADH), neurotoxicity (including peripheral neuropathy that may be reversible, leukoencephalopathy, and reversible posterior leukoencephalopathy syndrome [RPLS]), ocular toxicity (e.g., blurred vision, cerebral blindness, optic neuritis, papilledema), ototoxicity including tinnitus and hearing loss in the high-frequency range, peripheral neuropathy (may be irreversible), vascular toxicities (e.g., cerebral arteritis, CVA, MI, or thrombotic microangiopathy [hemolytic-uremic syndrome (HUS)]), and vestibular toxicity.
Overdose:
Deafness, intractable nausea and vomiting, kidney failure, liver failure, neuritis, ocular toxicity, significant myelosuppression, and death.
Antidote
Notify physician of all side effects. Cisplatin may have to be discontinued permanently or until recovery. Symptomatic and supportive treatment is indicated. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. Pretreatment with amifostine may reduce nephrotoxic, neurotoxic, and hematologic effects. Treat anaphylaxis with epinephrine, corticosteroids, oxygen, and antihistamines. There is no specific antidote. Hemodialysis appears to have little effect on removing platinum from the body because of the rapid and high degree of protein binding.
Cladribine 
(KLAD-rih-bean)
Leustatin
Antineoplastic (antimetabolite)
pH 6 to 6.6
Usual dose
In all situations, may be administered on an outpatient basis with an appropriate pump and a central venous line in place. Administer any subsequent course with extreme caution. Hematologic recovery must be considered.
Hairy cell leukemia:
0.09 mg/kg/day equally distributed as a continuous infusion over 24 hours. Repeat daily for 7 consecutive days.
Chronic lymphocytic leukemia and Waldenström’s macroglobulinemia (unlabeled):
0.1 mg/kg/day equally distributed as a continuous infusion over 24 hours for 7 consecutive days.
Acute myeloid leukemia and mantle cell lymphoma (unlabeled):
5 mg/M2/day over 2 hours for 5 days.
Dose adjustments
May be required with severe bone marrow impairment, with prior radiation or myelosuppressive agents. ■ May be required in severe renal insufficiency; effects of renal or hepatic impairment on excretion of cladribine not yet clarified for humans. ■ See Drug/Lab Interactions.
Dilution
Specific techniques required; see precautions.
Available in single-use 10-mL vials containing 10 mg (1 mg/mL). Contains no preservatives; aseptic technique imperative. May develop a precipitate at low temperatures. Warm naturally to room temperature and shake vigorously. Do not heat or microwave.
Inpatient continuous infusion:
Add the calculated daily dose of cladribine through a sterile 0.22-micron disposable hydrophilic syringe filter to an infusion bag containing 500 mL of NS.
Outpatient continuous infusion:
A total 7-day dose is added to a calculated amount of bacteriostatic NS to make a total volume of 100 mL. Add the calculated dose of cladribine (7 days × 0.09 mg/kg or 0.09 mL/kg) to the infusion reservoir through the sterile 0.22-micron disposable hydrophilic syringe filter, then add the calculated amount of bacteriostatic NS to the reservoir through the filter. Total volume in the reservoir should equal 100 mL. Specific equipment (i.e., a sterile medication reservoir and pump capable of delivering accurate minute amounts into a central venous line [presently using SIMS Deltec medication cassette with SIMS Deltec pump]) and a specific process including the use of a 0.22-micron syringe filter are required. Preparation of cassette usually done by pharmacist. Line of cassette remains clamped until attached to central venous line and pump is functional. See literature for details, and follow all specific instructions for medication pump.
Filters:
To minimize the risk of microbial contamination, a 0.22-micron hydrophilic syringe filter is required in the preparation of the outpatient continuous infusion.
Storage:
Protect from light. Refrigerate before reconstitution. Never refreeze. Discard any unused concentrate. 500 mL dilution is stable for at least 24 hours at RT under normal fluorescent light; may be refrigerated for up to 8 hours after dilution. Immediate use preferred. 100 mL dilution is stable in reservoir of medication cassette for 7 days if correctly diluted.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
D5W will cause degradation of cladribine. Manufacturer states, “Adherence to the recommended diluents and infusion systems is advised.”
One source suggests the following compatibilities:
Y-site:
Aminophylline, bumetanide, buprenorphine (Buprenex), butorphanol (Stadol), calcium gluconate, carboplatin (Paraplatin), chlorpromazine (Thorazine), cisplatin, cyclophosphamide (Cytoxan), cytarabine (ARA-C), dexamethasone (Decadron), diphenhydramine (Benadryl), dobutamine, dopamine, doxorubicin (Adriamycin), droperidol (Inapsine), enalaprilat (Vasotec IV), etoposide (VePesid), famotidine (Pepcid IV), furosemide (Lasix), gallium nitrate (Ganite), granisetron (Kytril), heparin, hydrocortisone sodium succinate (Solu-Cortef), hydromorphone (Dilaudid), idarubicin (Idamycin), leucovorin calcium, lorazepam (Ativan), mannitol, meperidine (Demerol), mesna (Mesnex), methylprednisolone (Solu-Medrol), metoclopramide (Reglan), mitoxantrone (Novantrone), morphine, nalbuphine, ondansetron (Zofran), paclitaxel (Taxol), potassium chloride (KCl), prochlorperazine (Compazine), promethazine (Phenergan), ranitidine (Zantac), sodium bicarbonate, teniposide (Vumon), vincristine.
Rate of administration
Inpatient continuous infusion:
A single dose properly diluted evenly distributed as an infusion over 24 hours.
Outpatient continuous infusion:
Administered through a central venous line (very concentrated solution). Medication reservoir and pump required (presently using Pharmacia Deltec medication cassette and pump worn as a portable pack). Set rate for equal distribution of 100 mL over 7 days. Follow all specific instructions for pump.
Actions
A chlorinated purine nucleoside analog and synthetic antineoplastic agent. Mechanism of action is not known, but it is believed to be cytotoxic by inhibiting both DNA synthesis and repair. Affects both dividing and resting cells. The 7-day course for hairy cell leukemia has resulted in complete response in a majority of patients with no evidence of persistent bone marrow disease. Crosses the blood-brain barrier. Average half-life is 4.2 to 9.2 hours. Specific methods of metabolism and routes of excretion are not known. Some drug does appear in urine.
Indications and uses
Treatment of active hairy cell leukemia (HCL) as defined by clinically significant anemia, neutropenia, thrombocytopenia, or disease-related symptoms.
Unlabeled uses:
Treatment of acute myeloid leukemia, chronic lymphocytic leukemia, mantle cell lymphoma, and Waldenström’s macroglobulinemia.
Contraindications
Hypersensitivity to cladribine or any of its components; neonates (7-day dilution contains benzyl alcohol).
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of the physician specialist. ■ Anticipate severe suppression of bone marrow function, including neutropenia, anemia, and thrombocytopenia; usually reversible and appears to be dose dependent. ■ Myelosuppressive effects are most notable during the first month after therapy. ■ Serious, sometimes fatal, infections have been reported (e.g., respiratory tract infections, pneumonia, viral skin infections, sepsis). ■ Neurologic toxicity, including paraparesis and quadriparesis, has been reported. Usually occurs with higher doses but has been seen with standard dosing regimens. ■ Because of the possibility of increased toxicity, use caution in known or suspected renal or hepatic insufficiency, or any severe bone marrow impairment, or prior cytoxic or radiation therapy. ■ Acute nephrotoxicity has been observed with high doses (4 to 9 times the recommended dose for HCL). Risk of toxicity increased when given concurrently with other nephrotoxic agents and/or therapies. ■ Rare cases of tumor lysis syndrome have been reported. ■ Appears to be no relationship between serum concentrations and ultimate clinical outcome. ■ Additional courses did not improve overall response. ■ Current studies suggest that overall response rate may be decreased in patients previously treated with splenectomy, deoxycoformycin (pentostatin), and in patients refractory to alpha-interferon. ■ May cause prolonged bone marrow hypocellularity; clinical significance not known.
Monitor:
Obtain baseline CBC with differential and platelets before therapy. May be repeated as indicated, but usually not required again until 7 or 8 days after treatment begins; then monitor as indicated for at least 4 to 8 weeks (anemia, neutropenia, thrombocytopenia, infection [bacterial, fungal, or viral], and bleeding are common and must be treated promptly). Monitoring schedule facilitates outpatient treatment; keep in close contact with patient. ■ Consider possibility of infection if fever occurs; appropriate lab tests, x-rays, and broad-spectrum antibiotics may be indicated, especially in a febrile neutropenic patient. ■ Monitor uric acid levels before and during treatment; maintain hydration; allopurinol may be indicated (preferred agent). Alkalinization of urine may also be indicated. ■ Monitor renal and hepatic function periodically. ■ Platelet count usually returns to normal in 12 days (may be delayed if severe baseline thrombocytopenia was present), absolute neutrophil count (ANC) usually returns to normal in 5 weeks, and hemoglobin in 8 weeks. All should be normal by 9 weeks. ■ Complete response is indicated by an absence of hairy cells in bone marrow and peripheral blood and normalization of peripheral blood parameters. Confirm response with bone marrow aspiration and biopsy between 9 weeks and 4 months. ■ Prophylactic antiemetics may improve patient comfort. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). Avoid constipation and avoid alcohol and aspirin (risk of GI bleeding).
Patient education:
Avoid pregnancy; consider birth control options and future fertility. ■ Report fever, bleeding, cough, edema, injection site reactions, malaise, mouth sores, rashes, shortness of breath, stomach pain, and tachycardia promptly. Maintain hydration. ■ Manufacturer supplies a patient education booklet; review thoroughly and discuss with physician and nurse. ■ Review all literature provided with pump to deliver outpatient dosing. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy; has potential to cause fetal harm. Has caused suppression of testicular cells in monkeys; effect on human fertility unknown. ■ Discontinue breast-feeding. ■ Safety for use in pediatric patients not established. Investigationally used in higher doses to treat relapsed acute leukemia. Dose-limiting toxicity occurred.
Elderly:
Geriatric-specific problems not encountered in studies to date. Consider age-related organ impairment.
Drug/lab interactions
Increased toxicity with other myelosuppressive agents (e.g., methotrexate). ■ May raise concentration of blood uric acid; increased doses of antigout agents (e.g., allopurinol [Aloprim]) may be indicated; avoid uricosurics (e.g., probenecid, sulfinpyrazone [Anturane]). ■ Do not administer live attenuated vaccines to patients receiving antineoplastic agents.
Side effects
Fever occurs first. Onset of thrombocytopenia begins in 7 to 10 days followed by anemia (severe) and neutropenia (severe). Fatigue, headache, injection site reactions, infection, nausea, and rash are common. Many other side effects may or may not be related to cladribine: abdominal pain, abnormal breath sounds, abnormal chest sounds, anorexia, arthralgia, asthenia, chills, constipation, cough, diaphoresis, diarrhea, dizziness, edema, epistaxis, erythema, insomnia, malaise, myalgia, pain, petechiae, pruritus, purpura, shortness of breath, tachycardia, trunk pain, weakness, vomiting.
Post-marketing:
Most of these additional side effects occurred in patients who received multiple courses of cladribine and include altered level of consciousness, aplastic anemia, conjunctivitis, elevated bilirubin and transaminases, hemolytic anemia, hypereosinophilia, hypersensitivity, myelodysplastic syndrome, neurologic toxicity, opportunistic infections, pulmonary interstitial infiltrates (usually with an infectious etiology), renal impairment, Stevens-Johnson syndrome, toxic epidermal necrolysis, and tumor lysis syndrome.
Overdose:
Acute nephrotoxicity, irreversible neurologic toxicity (paraparesis/quadriparesis), severe bone marrow suppression (anemia, neutropenia, and thrombocytopenia).
Antidote
Keep physician informed of all side effects; many will be treated symptomatically as indicated. Platelet or RBC transfusions are frequently required to treat anemia or thrombocytopenia, especially during the first month. Filgrastim (Neupogen, Zarxio) or pegfilgrastim (Neulasta) may be used to increase neutrophil count, although recovery is usually spontaneous. Use specific antibiotics to combat infection. Discontinue cladribine if renal toxicity, neurotoxicity, or overdose occurs. No specific antidote for overdose. Supportive therapy as indicated will help sustain the patient in toxicity. Resuscitate if indicated.
Clevidipine butyrate
(klev-ID-i-peen BUE-tih-rate)
Cleviprex
Calcium channel blocker
Antihypertensive
pH 6 to 8
Usual dose
Must be individualized to achieve the desired BP reduction. Titrate to patient response and BP goal.
Initial dose:
1 to 2 mg/hr (2 to 4 mL/hr) as a continuous infusion.
Dose titration:
Initially, double the dose at 90-second intervals. As the BP approaches goal, increase the dose by less than doubling and lengthen the time between dose adjustments to every 5 to 10 minutes. In general, a 1- to 2-mg/hr increase in dose will produce an additional 2- to 4-mm Hg decrease in systolic pressure. The maximum dose used for most patients in studies was 16 mg/hr.
Maintenance dose:
The desired therapeutic response for most patients occurs at 4 to 6 mg/hr (8 to 12 mL/hr). Doses as high as 32 mg/hr (64 mL/hr) have been administered. Data are limited. Because of lipid-load restrictions, no more than 1,000 mL or an average of 21 mg/hr (42 mL/hr) is recommended per 24-hour period. In studies, most infusions were administered for less than 24 hours. There is little experience with infusions lasting more than 72 hours at any dose.
Transition to oral therapy:
Discontinue or titrate the infusion downward while establishing appropriate oral therapy. Consider the lag time of onset of oral agent’s effect. See Monitor.
Dose adjustments
Lower-end initial doses may be indicated in the elderly. Consider the potential for decreased organ function and concomitant disease or drug therapy. ■ Patients with abnormal hepatic or renal function may receive the initial dose listed under Usual Dose.
Dilution
Strict aseptic technique is imperative. Available as a single-dose, ready-to-use vial containing a 0.5-mg/mL phospholipid emulsion that can support microbial growth. Invert vial gently several times before use to ensure uniformity of emulsion.
Filter:
No data available from manufacturer.
Storage:
Refrigerate unopened vials at 2° to 8° C (36° to 46° F) or store at 25° C (77° F) for up to 2 months. Vials stored at RT should not be returned to the refrigerator. Do not freeze. Protect from light until administration. Once vial is punctured, use within 4 hours and discard any unused portion, including that which is currently being infused.
Compatibility
Manufacturer states, “Should not be administered in the same line as other medications. Should not be diluted, but can be administered via a Y-tube or medication port with SWFI, NS, D5W, D5NS, D5LR, LR, and 10% amino acid.”
Rate of administration
Administer as a continuous infusion as outlined in Usual Dose. Use of an infusion device required. May be given through a central or peripheral line.
Actions
A dihydropyridine calcium channel blocker. Mediates the influx of calcium during depolarization in arterial smooth muscle. Reduces mean arterial BP by decreasing systemic vascular resistance in a dose-dependent manner. Does not reduce cardiac filling pressure (preload), confirming the lack of effect on venous capacitance vessels. Vasodilation and the resulting decrease in BP may produce a reflex increase in heart rate. Onset of effects begins within 2 to 4 minutes. Evidence of tolerance or hysteresis has not been observed in patients receiving infusions of up to 72 hours’ duration. Full recovery of BP is achieved 5 to 15 minutes after the infusion is stopped. Rapidly distributed. Highly protein bound. Metabolized by hydrolysis in the blood and extravascular tissues, making its elimination unlikely to be affected by hepatic or renal dysfunction. Half-life is approximately 15 minutes. Excreted primarily in urine and, to a lesser extent, in feces.
Indications and uses
Reduction of BP when oral therapy is not feasible or not desirable.
Contraindications
Allergies to soybeans, soy products, eggs, or egg products. ■ Defective lipid metabolism such as pathologic hyperlipidemia, lipoid nephrosis, or acute pancreatitis if accompanied by hyperlipidemia. ■ Severe aortic stenosis (afterload reduction can be expected to reduce myocardial oxygen delivery).
Precautions
For IV use only. ■ Strict aseptic technique required; see Dilution. ■ May produce systemic hypotension and reflex tachycardia. Dose reduction may be indicated. ■ Use caution in patients with lipid metabolism disorders. Contains approximately 0.2 Gm of lipid per mL. Lipid intake restrictions may be necessary in these patients. A reduction in the quantity of concurrently administered lipids (e.g., propofol, IV fat) may be necessary to compensate for the amount of lipid infused as part of the clevidipine formulation. See Usual Dose. ■ May produce a negative inotropic effect and exacerbate heart failure. ■ Rebound hypertension may occur in patients undergoing prolonged therapy; see Monitor. ■ Has not been studied for the treatment of hypertension associated with pheochromocytoma.
Monitor:
Monitor BP and HR during infusion and until vital signs are stable. ■ Rebound hypertension may occur in patients who receive a prolonged infusion and are not transitioned to other antihypertensive therapies. Monitor BP for at least 8 hours after the infusion is discontinued. ■ During transition to oral therapy, continue BP monitoring until patient is stabilized. ■ Monitor heart failure patients closely.
Patient education:
Promptly report signs of a hypertensive emergency (e.g., neurologic symptoms, vision changes, evidence of CHF). ■ Continued follow-up and treatment of pre-existing hypertension required.
Maternal/child:
Category C: use during pregnancy only if potential benefit justifies potential risk to the fetus. ■ Safety in labor and delivery not established. Other calcium channel blockers suppress uterine contractions in humans. ■ Safety for use in breast-feeding not established; effects unknown. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
Response similar to that seen in younger adults; however, greater sensitivity in the elderly cannot be ruled out. ■ See Dose Adjustments.
Drug/lab interactions
Formal studies have not been conducted. ■ Does not have the potential for inducing or inhibiting the cytochrome P450 system. ■ Use of beta-blockers as treatment for clevidipine-induced reflex tachycardia is not recommended. Experience is limited. ■ If used concomitantly with beta-blockers and beta-blockers are to be discontinued, withdraw beta-blocker therapy gradually. Clevidipine will not protect against the effects of abrupt beta-blocker withdrawal.
Side effects
Acute renal failure, atrial fibrillation, cardiac arrest, dyspnea, flushing, headache, hypotension, myocardial infarction, nausea, peripheral edema, rebound hypertension, reflex tachycardia, syncope, and vomiting.
Post-marketing:
Decreased oxygen saturation (possible pulmonary shunting), hypersensitivity reactions, increased blood triglycerides, ileus.
Antidote
Notify physician of any side effects; most will be treated symptomatically. Reduce dose for systemic hypotension or reflex tachycardia. Discontinue for suspected overdose. Reduction in antihypertensive effects should be seen within 5 to 15 minutes. Monitor BP and support if needed. Resuscitate as necessary.
Clindamycin phosphate 
(klin-dah-MY-cin FOS-fayt)
Cleocin Phosphate
Antibacterial
Antiprotozoal (lincosamide)
pH 5.5 to 7
Usual dose
Doses based on susceptibility of specific organisms; see literature.
Serious infections:
600 to 1,200 mg/24 hr in 2, 3, or 4 equally divided doses (300 to 600 mg every 12 hours, 200 to 400 mg every 8 hours, or 150 to 300 mg every 6 hours).
More severe infections:
1,200 to 2,700 mg/24 hr in 2, 3, or 4 equally divided doses (600 to 1,350 mg every 12 hours, 400 to 900 mg every 8 hours, or 300 to 675 mg every 6 hours).
Life-threatening infections:
Up to 4,800 mg/24 hr has been given.
Acute pelvic inflammatory disease (unlabeled):
900 mg every 8 hours. Used in combination with gentamicin. Continue both drugs for at least 24 hours after patient improves. Complete 14-day treatment program with oral doxycycline or clindamycin.
CNS toxoplasmosis in AIDS (unlabeled):
600 mg every 6 hours. Used in combination with pyrimethamine (Daraprim) and leucovorin.
Pneumocystis jiroveci pneumonia (unlabeled):
600 mg every 6 hours or 900 mg every 8 hours. Used in combination with primaquine.
Babesiosis (unlabeled):
1,200 to 2,400 mg/24 hr in 4 equally divided doses (300 to 600 mg every 6 hours). Continue for 7 to 10 days. Used in combination with quinine.
Prophylaxis of bacterial endocarditis:
600 mg IV or PO 30 minutes before procedure.
Pediatric dose
Pediatric patients over 1 month of age:
20 to 40 mg/kg of body weight/24 hr in 3 or 4 equally divided doses based on the seriousness of the infection (5 to 10 mg/kg every 6 hours or 6.66 to 13.3 mg/kg every 8 hours). Alternately 350 mg/M2/24 hr may be used (87.5 mg/M2 every 6 hours or 116.6 mg/M2 every 8 hours). 450 mg/M2/24 hr may be used for more serious infections if necessary (112.5 mg/M2 every 6 hours or 150 mg/M2 every 8 hours).
Prophylaxis of bacterial endocarditis:
20 mg/kg IV or PO 30 minutes before procedure. Do not exceed adult dose.
Neonatal dose
See Maternal/Child.
Under 1 month of age, full term:
3.75 to 5 mg/kg every 6 hours or 5 to 6.7 mg/kg every 8 hours. Another source suggests:
Neonates 7 days of age or less weighing less than or equal to 2 kg:
5 mg/kg every 12 hours.
Neonates 7 days of age or less weighing over 2 kg:
5 mg/kg every 8 hours.
Neonates over 7 days of age weighing less than 1.2 kg:
5 mg/kg every 12 hours.
Neonates over 7 days of age weighing 1.2 to 2 kg:
5 mg/kg every 8 hours.
Neonates over 7 days of age weighing over 2 kg:
5 mg/kg every 6 hours.
Dose adjustments
Dose adjustments are not required in the presence of mild to moderate renal or hepatic disease and should not be required in severe disease; see Monitor.
Dilution
Available prediluted in 300-, 600-, and 900-mg doses, in ADD-Vantage vials for use with ADD-Vantage infusion containers, as a 150 mg/mL solution in flip-top vials, in ready-to-use Galaxy bags, and in pharmacy bulk packages (for use only by the pharmacy). Dilute ADD-Vantage vials with 50 to 100 mL of D5W or NS. Doses prepared using the 150 mg/mL solutions are most commonly diluted in 50 to 100 mL of D5W, NS, or other compatible infusion solution. Concentration of clindamycin in diluent should not exceed 18 mg/mL. See chart on inside back cover or product insert for additional diluents. May be further diluted in larger amounts of compatible infusion solutions and given as a continuous infusion after the initial dose.
Storage:
Store vials at CRT before use. Administration as soon as possible after dilution is recommended. Stable at CRT for at least 24 hours in compatible infusion solutions. See manufacturer’s literature for additional stability data if diluted solutions are kept at RT, refrigerated, or frozen. Frozen solutions should be thawed at room temperature and not refrozen.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer lists as compatible for 24 hours at RT with IV solutions containing sodium chloride, glucose, calcium, or potassium and with solutions containing vitamin B complex in concentrations usually used clinically. Manufacturer states, “No incompatibility has been demonstrated with the antibiotics gentamicin, kanamycin, or penicillin.”
Manufacturer lists as incompatible with aminophylline, ampicillin, barbiturates, calcium gluconate, magnesium sulfate, phenytoin (Dilantin).
Sources suggest the following compatibilities:
Solution:
Isolyte H, D5/Isolyte M, D5/Isolyte P, Normosol R; see chart on inside back cover.
Additive:
Manufacturer states no incompatibility has been demonstrated with gentamicin or penicillin. Other sources list amikacin, ampicillin, aztreonam (Azactam), cefazolin (Ancef), cefepime (Maxipime), cefotaxime (Claforan), cefoxitin (Mefoxin), ceftazidime (Fortaz), cefuroxime (Zinacef), fluconazole (Diflucan), heparin, hydrocortisone sodium succinate (Solu-Cortef), methylprednisolone (Solu-Medrol), metoclopramide (Reglan), potassium chloride (KCl), ranitidine (Zantac), sodium bicarbonate, tobramycin, verapamil.
Y-site:
Acyclovir (Zovirax), amifostine (Ethyol), amiodarone (Nexterone), amphotericin B cholesteryl (Amphotec), anidulafungin (Eraxis), aztreonam (Azactam), bivalirudin (Angiomax), ceftaroline (Teflaro), cisatracurium (Nimbex), cyclophosphamide (Cytoxan), dexmedetomidine (Precedex), diltiazem (Cardizem), docetaxel (Taxotere), doxorubicin liposomal (Doxil), enalaprilat (Vasotec IV), esmolol (Brevibloc), etoposide phosphate (Etopophos), fenoldopam (Corlopam), fludarabine (Fludara), foscarnet (Foscavir), gemcitabine (Gemzar), granisetron (Kytril), heparin, hetastarch in electrolytes (Hextend), hydromorphone (Dilaudid), 6% hydroxyethyl starch (Voluven), labetalol, levofloxacin (Levaquin), linezolid (Zyvox), magnesium sulfate, melphalan (Alkeran), meperidine (Demerol), midazolam (Versed), milrinone (Primacor), morphine, multivitamins (M.V.I.), nicardipine (Cardene IV), ondansetron (Zofran), pemetrexed (Alimta), piperacillin/tazobactam (Zosyn), propofol (Diprivan), remifentanil (Ultiva), sargramostim (Leukine), tacrolimus (Prograf), teniposide (Vumon), theophylline, thiotepa, vinorelbine (Navelbine), zidovudine (AZT, Retrovir).
Rate of administration
Should not be administered intravenously undiluted as a bolus. Severe hypotension and cardiac arrest can occur with too-rapid injection.
Intermittent infusion:
30 mg or fraction thereof over at least 1 minute (each 300 mg over a minimum of 10 minutes/1,200 mg over a minimum of 40 to 60 minutes). Do not give more than 1,200 mg in single 1-hour infusion.
Continuous infusion:
Administer initial dose at 10 (15 or 20) mg/min over 30 minutes (rapid infusion rate). Will result in serum levels above 4 (5 or 6) mg/mL. To maintain these serum levels, continue infusion at 0.75 (1 or 1.25) mg/min.
Actions
A semi-synthetic antibiotic that quickly converts to active clindamycin. Bacteriostatic with activity against gram-positive aerobes and anaerobes, as well as some gram-negative anaerobes. It inhibits bacterial protein synthesis by binding to the 50S subunit of the ribosome. Widely distributed in most body fluids and tissues. There is no clinically effective distribution to cerebrospinal fluid. Half-life is approximately 3 hours. Excreted in urine and feces in small amounts. Crosses placental barrier. Secreted in breast milk.
Indications and uses
Treatment of serious infections caused by susceptible anaerobic bacteria; treatment of infections due to susceptible strains of streptococcal, pneumococcal, and staphylococcal bacteria in penicillin-allergic patients; or treatment of infections that do not respond or are resistant to other less toxic antibiotics, such as erythromycin. Conditions indicated include septicemia, lower respiratory tract infections (including pneumonia), and skin and skin structure, gynecologic, intra-abdominal, and bone and joint infections.
Unlabeled uses:
Alternative to sulfonamides with pyrimethamine to treat CNS toxoplasmosis in AIDS patients. ■ Treat Pneumocystis jiroveci pneumonia in combination with primaquine. ■ Treatment of babesiosis. ■ Prophylaxis of bacterial endocarditis. ■ Treatment of acute pelvic inflammatory disease.
Contraindications
Known hypersensitivity to clindamycin or lincomycin.
Precautions
To reduce the development of drug-resistant bacteria and maintain its effectiveness, clindamycin should be used to treat or prevent only those infections proven or strongly suspected to be caused by bacteria. ■ Sensitivity studies are indicated to determine susceptibility of the causative organism to clindamycin. ■ Avoid prolonged use; superinfection caused by overgrowth of nonsusceptible organisms may result. ■ Because its use has been associated with severe colitis, it should be reserved for serious infections in which less toxic antimicrobial agents are inappropriate. ■ Clostridium difficile–associated diarrhea (CDAD) has been reported. May range from mild diarrhea to fatal colitis. Consider in patients who present with diarrhea during or after treatment with clindamycin. ■ Use caution with a history of GI, severe renal, or liver disease, and in patients with a history of asthma or significant allergies. ■ Severe skin reactions such as toxic epidermal necrolysis, some with fatal outcome, have been reported. ■ Serious anaphylactoid reactions have been reported. ■ Certain infections may require incision and drainage or other indicated surgical procedures in addition to antibiotic therapy. ■ Not appropriate to treat meningitis.
Monitor:
Capable of causing severe, even fatal, colitis; observe for symptoms of diarrhea. ■ Periodic blood cell counts and liver and kidney studies are indicated in prolonged therapy. ■ Monitor liver enzymes periodically in patients with severe liver disease. ■ Monitor for S/S of hypersensitivity reactions, including severe skin and anaphylactoid reactions.
Patient education:
Promptly report diarrhea or bloody stools that occur during treatment or up to several months after an antibiotic has been discontinued; may indicate CDAD and require treatment. ■ Do not treat diarrhea without notifying physician.
Maternal/child:
Category B: in clinical trials with pregnant women, the systemic administration of clindamycin during the second or third trimesters has not been associated with an increased frequency of congenital abnormalities. Should be used during the first trimester only if clearly needed. ■ Discontinue breast-feeding. ■ Contains benzyl alcohol, which has been associated with a fatal “gasping syndrome” in neonates. ■ Benzyl alcohol can cross the placenta. ■ Monitor organ system functions if used in pediatric patients.
Elderly:
CDAD may occur more frequently in elderly patients (greater than 60 years of age) and may be more severe. Monitor carefully for changes in bowel frequency; may not tolerate diarrhea well.
Drug/lab interactions
May potentiate neuromuscular blocking agents (e.g., atracurium [Tracrium]) and cause profound respiratory depression. ■ Antagonized by erythromycin. ■ May decrease cyclosporine (Sandimmune) levels. Monitor and adjust dose if necessary.
Side effects
Abdominal pain, abnormal liver function tests, agranulocytosis, anaphylaxis, azotemia, cardiac arrest, CDAD, diarrhea, drug reaction with eosinophilia and systemic symptoms (DRESS), eosinophilia (transient), esophagitis, hypersensitivity reactions, hypotension, injection site reactions, jaundice, leukopenia, metallic taste, nausea, neutropenia (transient), oliguria, polyarthritis (rare), proteinuria, pruritus, pseudomembranous colitis, severe skin reactions (e.g., toxic epidermal necrolysis, acute generalized exanthematous pustulosis [AGEP], erythema multiforme), skin rashes, thrombophlebitis, urticaria, vaginitis, vomiting.
Antidote
Notify the physician of any side effects. Discontinue the drug if indicated (e.g., CDAD, diarrhea, hypersensitivity reactions, severe skin reaction), treat hypersensitivity reactions as indicated, and resuscitate as necessary. Mild cases of CDAD may respond to discontinuation of drug. Treat CDAD with fluids, electrolytes, protein supplements, and oral vancomycin (Vancocin) or metronidazole (Flagyl) as indicated. In severe cases, surgical evaluation may be indicated. Hemodialysis or CAPD will not decrease blood levels in toxicity.
Clofarabine
(kloh-FARE-ah-bean)
Clolar
Antineoplastic (metabolic inhibitor)
pH 4.5 to 7.5
Pediatric dose
Premedication:
Consider prophylactic antiemetic medications. Consider use of prophylactic steroids (e.g., hydrocortisone 100 mg/M2 on Days 1 through 3) to mitigate or prevent the development of systemic inflammatory response syndrome (SIRS) or capillary leak syndrome (e.g., hypotension, pulmonary edema, tachycardia, tachypnea). See Drug/Lab Interactions.
Clofarabine:
52 mg/M2 as an infusion each day for 5 consecutive days. Dose is based on body surface area (BSA), which is calculated using the actual height and weight before the start of each cycle. Repeat every 2 to 6 weeks; see Dose Adjustments. Frequency is based on recovery or return to baseline organ function. Median time between cycles during clinical studies was 28 days (range 12 to 55 days).
Dose adjustments
Administer subsequent cycles no sooner than 14 days from the starting day of the previous cycle provided the patient’s ANC is greater than or equal to 0.75 × 109/L. ■ Reduce dose by 50% in patients with a CrCl between 30 and 60 mL/min. Information is insufficient to make a dose recommendation in patients with a CrCl less than 30 mL/min or in patients on dialysis. ■ Reduce the dose of the next cycle by 25% in patients experiencing a Grade 4 neutropenia (ANC less than 0.5 × 109/L) that lasts 4 or more weeks. ■ Withhold clofarabine if a clinically significant infection develops. When the infection is clinically controlled, restart therapy at full dose. ■ Discontinue if hypotension develops at any time during the 5 days of administration. ■ Discontinue drug immediately if Grade 3 or higher increases in SCr, liver enzymes, or bilirubin occur; see Monitor and Antidote. May be restarted (generally with a 25% dose reduction) when the patient is stable and organ function has returned to baseline. ■ Discontinue drug immediately if S/S of SIRS or capillary leak syndrome occur; see Monitor and Antidote. May be restarted (generally with a 25% dose reduction) when the patient is stable. ■ Withhold clofarabine if a Grade 3 noninfectious nonhematologic toxicity occurs (excluding transient elevations in serum transaminases and/or serum bilirubin and/or nausea and vomiting that is controlled by antiemetics). With resolution or a return to baseline, restart clofarabine at a 25% dose reduction. ■ Discontinue therapy if a Grade 4 noninfectious nonhematologic toxicity occurs.
Dilution
Specific techniques required; see precautions.
Available in single-use 20-mL vials containing 20 mg (1 mg/mL). Contains no preservatives; aseptic technique is imperative.
Calculate the exact number of vials needed to achieve the total dosing volume required.
Total number of vials required = Total dose (mg) ÷ 20 mg
Dosing volume (mL) = Total dose (mg)
For example, a child with a body surface area (BSA) of 0.75 M2 would need a dose of 39 mg of clofarabine (39 ÷ 20 mg = 1.95 vials, so 2 vials of clofarabine would be needed; 39 mg equals a dosing volume of 39 mL).
Withdraw the calculated dose from the vial(s). Using a 0.2-micron syringe filter, add the calculated dose to a sufficient volume of D5W or NS to provide a final concentration between 0.15 mg/mL and 0.4 mg/mL (e.g., 39 mg [39 mL in the example above] added to 100 mL of D5W or NS will provide a final concentration of approximately 0.28 mg/mL).
Filters:
Use of a 0.2-micron syringe filter is recommended for use during dilution in D5W or NS.
Storage:
Store unopened vials at CRT. Diluted solutions may be stored at CRT but must be used within 24 hours.
Compatibility
Manufacturer states, “To prevent drug incompatibilities, no other medications should be administered through the same IV line.”
Rate of administration
A single dose properly diluted and evenly distributed as an infusion over 2 hours.
Actions
A purine nucleoside metabolic inhibitor. Acts by inhibiting DNA synthesis. Also disrupts the mitochondrial membrane, causing the release of mitochondrial proteins, cytochrome C, and apoptosis-inducing factor, which leads to cell death. Is cytotoxic to rapidly proliferating and quiescent cancer cell types in vitro. Results in a rapid reduction of peripheral leukemia cells. Metabolism in the liver is very limited. Pathways of nonhepatic elimination not known. Estimated half-life is 5.2 hours. Excreted primarily in the urine.
Indications and uses
Treatment of pediatric patients 1 to 21 years of age with relapsed or refractory acute lymphoblastic leukemia (ALL) after treatment with at least two prior regimens. This indication is based on response rate. There are no trials verifying an improvement in disease-related symptoms or increased survival with clofarabine.
Unlabeled uses:
Treatment of other relapsed or refractory leukemias, including acute myelocytic leukemia (AML), myelodysplastic syndrome (MDS), and chronic myeloid leukemia in blast phase.
Contraindications
Manufacturer states, “None.”
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of the physician specialist in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ At the initiation of treatment, most patients have hematologic impairment as a manifestation of the leukemia. Clofarabine causes myelosuppression, which may be severe and prolonged. Treatment may result in prolonged and severe neutropenia, including febrile neutropenia. Patients may be at an increased risk for infection, including severe and fatal sepsis and opportunistic infections. ■ Use with great caution in patients with impaired hepatic or renal function. ■ May develop tumor lysis syndrome, cytokine release syndrome, systemic inflammatory response syndrome, capillary leak syndrome, and organ dysfunction; deaths have been reported; see Monitor. ■ Serious and fatal hemorrhage, including cerebral, GI, and pulmonary hemorrhage, has occurred. Most cases were associated with thrombocytopenia; see Monitor. ■ Patients who have previously received a hematopoietic stem cell transplant (HSCT) are at higher risk for hepatotoxicity suggestive of veno-occlusive disease (VOD) following treatment with clofarabine (40 mg/M2) used in combination with etoposide (VePesid 100 mg/M2) and cyclophosphamide (Cytoxan 440 mg/M2); has also been reported with monotherapy. ■ Severe and fatal hepatotoxic events, including hepatitis and hepatic failure, have been reported; see Monitor. ■ Renal toxicity, including Grade 3 or 4 elevated creatinine, acute renal failure, and hematuria, have been reported; see Monitor and Dose Adjustments. ■ Fatal and serious cases of enterocolitis (neutropenic colitis, cecitis, and C. difficile colitis) have been reported. Occurs more frequently within 30 days of treatment and in the setting of combination chemotherapy. May lead to necrosis, perforation, hemorrhage, or sepsis complications. ■ Serious and fatal skin reactions (Stevens-Johnson syndrome [SJS] and toxic epidermal necrolysis [TEN]) have been reported. ■ Safety and effectiveness in adults not established. In a Phase 1 study of adults with refractory and/or relapsed hematologic malignancies, the pediatric dose of 52 mg/M2 was not tolerated.
Monitor:
Obtain baseline CBC with differential and platelets and serum electrolytes. Bone marrow suppression is expected. Appears to be dose-dependent and is usually reversible (with interruption of clofarabine treatment) but can be severe; monitor regularly and more frequently in patients who develop cytopenias. Monitoring of CBC and platelets is recommended daily during the 5 days of clofarabine administration, then 1 to 2 times weekly or as clinically indicated. ■ Obtain baseline renal and hepatic function studies (e.g., SCr, bilirubin, ALT, AST), and uric acid levels. Monitor renal and hepatic function closely during the 5 days of clofarabine administration. Monitor for S/S of hepatitis and hepatic failure. Discontinue drug immediately if Grade 3 or higher increases in SCr, liver enzymes, or bilirubin occur; see Antidote. ■ Monitor HR, BP, and respiratory status closely during infusion. Hypotension should be reported immediately; see Antidote. ■ Monitor for S/S of tumor lysis syndrome (e.g., hyperkalemia, hyperphosphatemia, hyperuricemia, hypocalcemia, metabolic acidosis, urate crystalluria, and renal failure). Adequate hydration, antihyperuricemics (e.g., allopurinol), and alkalinization of urine are indicated to prevent and/or treat hyperuricemia due to tumor lysis syndrome. ■ Monitor for S/S of cytokine release syndrome (e.g., hypotension, pulmonary edema, tachycardia, tachypnea); may develop into systemic inflammatory response syndrome (SIRS)/capillary leak syndrome (rapid onset of respiratory distress, hypotension, pleural and pericardial effusion, and multiorgan failure). Close monitoring and early intervention may reduce risk; see Pediatric Dose and Antidote. To reduce the effects of tumor lysis and other adverse events (e.g., SIRS), the continuous administration of IV fluids throughout the 5 days of clofarabine treatment is recommended. ■ Observe closely for signs of infection. Prophylactic antibiotics may be indicated pending the result of C/S in a febrile neutropenic patient. ■ Use prophylactic antiemetics to reduce nausea and vomiting and increase patient comfort. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Monitor patients for S/S of enterocolitis and treat promptly. ■ Monitor for development of skin reactions.
Patient education:
Avoid pregnancy; nonhormonal birth control recommended for men and women. Report a suspected pregnancy immediately. ■ Drink plenty of fluids and avoid dehydration that may be caused by diarrhea and vomiting; report promptly if significant. ■ Laboratory monitoring required. ■ Promptly report bleeding, decreased urine output, dizziness, fainting spells, infection, jaundice, light-headedness, rapid respiratory rate, or a rapid heart rate. ■ Review medications with pharmacist or physician. Avoid medications that may be hepatotoxic or nephrotoxic, including OTC or herbal medications. ■ Skin rash may occur; report promptly if significant. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy; may cause fetal harm. Also has dose-related effects on male reproductive organs in animals. ■ Discontinue breast-feeding.
Elderly:
Not indicated in this patient population.
Drug/lab interactions
Clinical drug-drug interactions have not been studied; however, the following cautions should be considered. ■ Primarily excreted by the kidneys; minimize exposure to drugs with known renal toxicity during the 5 days of clofarabine administration (e.g., aminoglycosides [e.g., gentamicin], amphotericin B, NSAIDs [e.g., ibuprofen (Advil, Motrin), naproxen (Aleve, Naprosyn)], rifampin [Rifadin]). Risk of renal toxicity may be increased. ■ The liver is a known target organ for toxicity; consider avoiding drugs known to induce hepatic toxicity (e.g., amiodarone [Nexterone], NSAIDs [e.g., ibuprofen (Advil, Motrin), naproxen (Aleve, Naprosyn)], phenothiazines [e.g., prochlorperazine (Compazine)], zidovudine [AZT, Retrovir]). ■ Close monitoring is required with concomitant administration of medications affecting blood pressure or cardiac function (e.g., diuretics [furosemide (Lasix)], calcium channel blockers [diltiazem (Cardizem)], and other antihypertensives). ■ Cytochrome P450 inhibitors (e.g., cimetidine [Tagamet], erythromycins, antifungal agents [e.g., itraconazole (Sporanox)], ritonavir [Norvir], verapamil) and cytochrome P450 inducers (e.g., carbamazepine [Tegretol], phenobarbital, phenytoin [Dilantin], rifampin [Rifadin]) are unlikely to affect the metabolism of clofarabine. The effect on cytochrome P450 substrates has not been studied.
Side effects
Bone marrow suppression (e.g., anemia, leukopenia, neutropenia, thrombocytopenia) is anticipated, appears to be dose dependent, and is usually reversible. Anxiety, diarrhea, fatigue, febrile neutropenia, fever, flushing, headache, mucosal inflammation, nausea and vomiting, palmar-plantar erythrodysesthesia syndrome, pruritus, and rash occur most frequently. SIRS/capillary leak syndrome (e.g., rapid onset of respiratory distress, hypotension, pleural and pericardial effusion, and multiorgan failure) has occurred and can be fatal. Hepatotoxicity (elevated AST, ALT, bilirubin), renal toxicity (elevated SCr, acute renal failure), and veno-occlusive disease of the liver have occurred. Serious and fatal hemorrhage (cerebral, GI, and pulmonary), enterocolitis (neutropenic colitis, cecitis, and C. difficile colitis), and skin reactions (SJS and TEN) have been reported. Numerous additional side effects may occur and include abdominal pain, anorexia, arthralgia, back pain, cardiac toxicity (e.g., pericardial effusion, tachycardia), confusion, constipation, cough, depression, dermatitis, dizziness, dyspnea, edema, elevated creatinine, epistaxis, erythema, gingival bleeding, hematuria, hepatobiliary toxicity (e.g., elevated AST, ALT, bilirubin), hepatomegaly, hypertension, hypotension, infections (e.g., bacteremia, cellulitis, herpes simplex, oral candidiasis, pneumonia, sepsis, staphylococcal), injection site pain, irritability, jaundice, lethargy, myalgia, pain, petechiae, pleural effusion, renal toxicity, respiratory distress, rigors, somnolence, sore throat, transfusion reaction, tremor, weight loss.
Post-marketing:
Bone marrow failure, GI hemorrhage (including fatalities), hepatic failure, hepatitis, hyponatremia, Stevens-Johnson syndrome, toxic epidermal necrolysis, and veno-occlusive disease.
Antidote
Keep physician informed of all side effects; most will be treated symptomatically as indicated. Discontinue if hypotension develops at any time during the 5 days of administration. Discontinue clofarabine immediately if early S/S of SIRS or capillary leak (e.g., hypotension) appear. Use of albumin, diuretics, and steroids may be indicated. May consider restarting (usually at a lower dose) after the patient is stabilized and organ function has returned to baseline. Discontinue clofarabine immediately if substantial increases in SCr, liver enzymes, or bilirubin occur. May be restarted (possibly at a lower dose) when the patient is stable and organ function has returned to baseline. Discontinue clofarabine for exfoliative or bullous rash or if SJS or TEN is suspected. Bone marrow suppression must be resolved before additional doses can be given. Administration of whole blood products (e.g., packed RBCs, platelets, or leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. Use specific antibiotics to combat infection. No specific antidote for overdose. Supportive therapy as indicated will help sustain the patient in toxicity. Should a hypersensitivity reaction occur, treat with antihistamines, corticosteroids, epinephrine, and oxygen as indicated. Resuscitate if indicated.
Coagulation factor VIIa (recombinant) RTS 
(ko-ag-yew-LA-shun FAK-ter 7a [re-KOM-be-nant])
NovoSeven RT
Antihemorrhagic
pH 5.5
Usual dose
Hemophilia A or B patients with inhibitors
Bleeding episodes:
90 mcg/kg every 2 hours until hemostasis is achieved or until the treatment has been judged to be ineffective. Doses between 35 and 120 mcg/kg have been used successfully. Minimum effective dose has not been established. The dose and dosing interval may be adjusted based on the severity of the bleeding and the degree of homeostasis achieved. In clinical studies, a decision on outcome was reached for a majority of patients with joint or muscle bleeds within 8 doses, although more doses were required for severe bleeds. For severe bleeds, dosing should continue at 3- to 6-hour intervals after hemostasis is achieved to maintain the hemostatic plug. The appropriate duration of posthemostatic dosing has not been studied and should be minimized; see Precautions. If a new bleeding episode or rebleeding occurs, return to 2-hour dosing intervals.
Surgical intervention:
90 mcg/kg immediately before the intervention. Repeat every 2 hours during intervention. For minor surgery, postsurgical dosing should be administered every 2 hours for 48 hours and then every 2 to 6 hours until healing has occurred. For major surgery, postsurgical dosing should be administered every 2 hours for 5 days and then every 4 hours until healing has occurred. Additional doses may be given if required.
Congenital factor VII deficiency patients
Bleeding episodes and surgical intervention:
15 to 30 mcg/kg every 4 to 6 hours until hemostasis is achieved. Doses as low as 10 mcg/kg have been effective. Dose and dosing interval should be adjusted to each individual based on the severity of bleeding and the degree of hemostasis achieved. Minimal effective dose has not been determined.
Acquired hemophilia
70 to 90 mcg/kg every 2 to 3 hours until hemostasis is achieved. Minimum effective dose has not been determined.
Pediatric dose
See Usual Dose. Clinical studies were conducted with dosing determined according to body weight and not according to age. See Maternal/Child.
Dose adjustments
Dose and administration interval may be adjusted based on the severity of the bleeding and the degree of hemostasis achieved. If patient develops intravascular coagulation or thrombosis, dosage should be reduced or treatment stopped; see Monitor.
Dilution
NovoSeven RT is room temperature stable (RTS); aseptic technique is imperative. Available in packages that contain 1-, 2-, or 5-mg vials with a specified volume of histidine diluent. Select the appropriate vial package based on the calculated dose. Bring vial and diluent to RT. Do not exceed 37° C (98.6° F). Reconstitute powder with provided diluent, aiming the needle (20- to 26-gauge needle recommended) and the stream of diluent against the side of the vial. Do not inject the diluent directly on the powder. Do not use SWFI or other diluents. Gently swirl vial until powder is completely dissolved. Final concentration of reconstituted solution is 1 mg/mL (1,000 mcg/mL).
Storage:
Before reconstitution, refrigerate or store between 2° and 25° C (36° and 77° F). Avoid exposure to direct sunlight. Do not freeze. After reconstitution, store at RT or refrigerate. Should be used within 3 hours. Do not freeze reconstituted product or store in syringe. Discard unused product.
Compatibility
Manufacturer states, “Intended for IV injection only and should not be mixed with infusion solutions. Do not store reconstituted solution in syringes.” If line needs to be flushed before or after NovoSeven RT administration, use NS.
Rate of administration
A single dose as a slow IV injection over 2 to 5 minutes, depending on dose administered.
Actions
A vitamin K–dependent glycoprotein structurally similar to human plasma–derived factor VIIa. Produced by recombinant DNA technology. Promotes hemostasis by activating the extrinsic pathway of the coagulation cascade. When complexed with tissue factor, can activate coagulation factor X to factor Xa, and coagulation factor IX to factor IXa. Factor Xa, in complex with other factors, then converts prothrombin to thrombin. This leads to the formation of a hemostatic plug by converting fibrinogen to fibrin. Half-life is 2.3 hours. Duration of action is 3 hours.
Indications and uses
Treatment of bleeding episodes or prevention of bleeding in surgical interventions or invasive procedures in hemophilia A or B patients with inhibitors to factor VIII or factor IX and in patients with acquired hemophilia. ■ Treatment of bleeding episodes or prevention of bleeding in surgical interventions or invasive procedures in patients with congenital FVII deficiency.
Contraindications
Manufacturer states, “None.” Use with caution in patients with known hypersensitivity to coagulation factor VIIa (recombinant), any of its components, and in patients with known hypersensitivity to mouse, hamster, or bovine proteins.
Precautions
For intravenous bolus administration only. ■ Should be administered to patients only under the direct supervision of a physician experienced in the treatment of bleeding disorders. ■ Thrombotic events have been reported in clinical trials as well as through post-marketing surveillance following coagulation factor VIIa (recombinant) RTS use for each of the approved indications. Patients with disseminated intravascular coagulation (DIC), advanced atherosclerotic disease, crush injury, septicemia, or concomitant treatment with aPCCs/PCCs (activated or nonactivated prothrombin complex concentrates) may have an increased risk of developing thrombotic events due to circulating tissue factor or predisposing coagulopathy. Use caution in patients with an increased risk of thromboembolic complications. These include, but are not limited to, patients with a history of coronary artery disease (CAD), liver disease, DIC, or postoperative immobilization; elderly patients; and neonates. ■ Coagulation factor VIIa has been studied in placebo-controlled trials outside the approved indications to control bleeding in intracranial hemorrhage, advanced liver disease, trauma, cardiac surgery, spinal surgery, and other therapeutic areas. Safety and effectiveness have not been established in these settings, and the use is not approved by the FDA. Arterial and venous thrombotic and thromboembolic events have been reported during post-marketing surveillance. Studies have shown an increased risk of arterial thromboembolic adverse events (e.g., MI, myocardial ischemia, cerebral infarction, and cerebral ischemia) with coagulation factor VIIa when administered outside the current approved guidelines. ■ Biologic and clinical effects of prolonged elevated levels of factor VIIa have not been studied; therefore, the duration of posthemostatic dosing should be minimized, and patients should be appropriately monitored by a physician experienced in the treatment of hemophilia during this time period.
Monitor:
Evaluation of hemostasis should be used to determine the effectiveness of therapy and to provide a basis for modification of the treatment schedule; coagulation parameters do not necessarily correlate with or predict the effectiveness of therapy. Coagulation parameters (e.g., PT, aPTT, plasma FVII clotting activity [FVII:C]) may be used as an adjunct to the clinical evaluation of hemostasis in monitoring the effectiveness and treatment schedule, although these parameters have shown no direct correlation to achieving hemostasis. ■ Patients with factor VII deficiency should be monitored for PT and factor VII coagulant activity before and after treatment. If the factor VIIa activity fails to reach the expected level, if PT is not corrected, or if bleeding is not controlled after treatment with the recommended doses, antibody formation should be suspected and analysis for antibodies should be performed. ■ The normal factor VII plasma concentration is 0.5 mcg/mL. Factor VIIa levels of 15% to 25% (0.075 to 0.125 mcg/mL) are generally sufficient to achieve normal hemostasis. ■ Monitor patients for the development of signs or symptoms of activation of the coagulation system or thrombosis. When there is laboratory confirmation of intravascular coagulation or presence of clinical thrombosis, dosage should be reduced or the treatment stopped, depending on the patient’s symptoms; see Dose Adjustments.
Patient education:
Discuss benefits versus risk of therapy and signs of hypersensitivity reactions including hives, urticaria, chest tightness, wheezing, hypotension, and anaphylaxis. ■ Signs of bleeding may be similar to signs of thrombosis and can include new-onset swelling and pain in the limbs or abdomen, new-onset chest pain, shortness of breath, loss of sensation or motor power, or altered consciousness or speech.
Maternal/child:
Category C: safety for use during pregnancy not established. Use only if clearly indicated and benefit justifies potential risk to the fetus. ■ Discontinue breast-feeding. ■ Thrombotic events have been reported in women without a prior diagnosis of bleeding disorders who have received coagulation factor VIIa for uncontrolled postpartum hemorrhage. ■ A decision should be made whether to discontinue nursing or to discontinue the drug. ■ The safety and effectiveness have not been studied to determine if there are differences among various age-groups from infants to adolescents (0 to 16 years of age); see Pediatric Dose.
Elderly:
Numbers in clinical studies insufficient to determine if the elderly respond differently than younger subjects.
Drug/lab interactions
The risk of potential interaction between factor VIIa and coagulation factor concentrates has not been adequately evaluated. Simultaneous use of activated prothrombin complex concentrates (e.g., anti-inhibitor coagulant complex [Feiba]) or prothrombin complex concentrates (e.g., factor IX [AlphaNine SD, Benefix]) should be avoided.
Side effects
Generally well tolerated. The majority of patients reporting side effects received more than 12 doses. The most common side effects are arthralgia, edema, fever, headache, hemorrhage, hypertension, hypotension, injection site reaction, nausea, pain, rash, and vomiting. Most serious adverse reactions are thrombotic events; however, the risk in patients with hemophilia and inhibitors is considered to be low. Fatal and nonfatal thrombotic events have been reported when used for both labeled and off-label indications. Other side effects include arthrosis, bradycardia, coagulation disorder, DIC, decreased fibrinogen plasma, decreased prothrombin, decreased therapeutic response, hemarthrosis, hypersensitivity reactions, increased fibrinolysis, pneumonia, pruritus, purpura, renal function abnormalities, shock, subdural hematoma, thrombosis, urticaria.
Post-marketing:
High d-dimer levels and consumptive coagulopathy; thromboembolic events, including myocardial ischemia and/or infarction, cerebral ischemia and/or infarction, thrombophlebitis, arterial thrombosis, deep vein thrombosis, and related pulmonary embolism; and isolated cases of hypersensitivity reactions (including anaphylaxis) have occurred following use in both labeled and unlabeled indications.
Antidote
Discontinue drug and notify physician of any major side effects. Treat hypersensitivity reactions as indicated. For thrombosis or DIC, anticoagulation with heparin may be indicated.
Coagulation factor X (human)
(ko-ag-yew-LA-shun FAK-ter X)
Coagadex
Antihemorrhagic
Usual dose (international units [IU])
Dose, frequency, and duration must be individualized based on the severity of the factor X deficiency, location and extent of bleeding, patient’s clinical condition, and individual clinical response. Each vial is labeled with the actual factor X potency/content in international units (IU). The dose to achieve a desired in vivo peak increase in factor X level can be calculated with the following formula.
Dose (IU) = Body weight (kg) × Desired factor X rise (IU/dL) × 0.5
The desired factor X rise is the difference between the patient’s plasma factor X level and the desired level. The dosing formula is based on the observed recovery of 2 IU/dL per IU/kg (i.e., for each 1 IU/kg of factor X administered, the circulating factor X level increases by approximately 2 IU/dL).
On-demand treatment and control of bleeding episodes:
25 IU/kg at the first sign of bleeding. Repeat every 24 hours until the bleed stops. Do not administer more than 60 IU/kg daily.
Perioperative management of bleeding:
Do not administer more than 60 IU/kg daily.
Presurgery:
Calculate the dose of coagulation factor X required to raise plasma factor X levels to 70 to 90 IU/dL using the following formula.
Required dose (IU) = Body weight (kg) × Desired factor X rise (IU/dL) × 0.5
Postsurgery:
Repeat dose as necessary to maintain plasma factor X levels at a minimum of 50 IU/dL until there is no longer a risk of bleeding due to surgery.
Dose adjustments
Adjust dose and duration based on factor X levels, location and extent of bleeding, patient’s clinical condition, and clinical response to therapy.
Dilution
Supplied in single-use glass vials containing approximately 250 or 500 IU (approximately 100 IU/mL after reconstitution) of factor X activity, packaged with 2.5 or 5 mL of SWFI, respectively, and a Mix2Vial transfer device. The total number of IUs available is clearly marked on each vial. Record the batch number of each vial. Consult instructions for reconstitution and administration in the package insert. If stored in the refrigerator, warm to room temperature (25° C) before reconstitution. If more than 1 vial is required, use a new Mix2Vial transfer set for each vial of drug. Solution from multiple vials can be pooled into a single syringe. Do not shake. Do not use if the reconstituted solution is cloudy or contains any particles. The reconstituted solution should be clear or a slightly pearl-like solution.
Filters:
Storage:
Store in refrigerator or at RT (2° to 30° C [36° to 86° F]) in original carton to protect from light until ready to use. Do not freeze. Do not use beyond the expiration date on the product vial. Should be used immediately but must be used within 1 hour of reconstitution.
Compatibility
Specific information not available; consider specific use and contact pharmacist.
Rate of administration
A single dose may be administered as an infusion at a rate of 10 mL/min up to a maximum rate of 20 mL/min; consider patient comfort. Reduce rate of administration or interrupt the infusion if a marked increase in pulse occurs.
Actions
Coagulation factor X is a plasma-derived, purified concentrate of human coagulation factor X. It temporarily replaces the missing factor X needed for effective hemostasis. After conversion to its active form (factor Xa), it associates with factor Va to form the prothrombinase complex, which activates prothrombin to thrombin. Thrombin then acts on soluble fibrinogen and factor XIII to generate a cross-linked fibrin clot. Mean half-life is approximately 30.3 hours.
Indications and uses
Treatment of adults and pediatric patients (12 years of age and above) with hereditary factor X deficiency for on-demand treatment and control of bleeding episodes and perioperative management of bleeding in patients with mild hereditary factor X deficiency.
Limitation of use:
Perioperative management of bleeding in major surgery in patients with moderate and severe hereditary factor X deficiency has not been studied.
Contraindications
Life-threatening hypersensitivity reactions to coagulation factor X or any of the product components.
Precautions
For IV use only. ■ Administered under the direction of a physician knowledgeable in the treatment of coagulation disorders in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Hypersensitivity reactions, including anaphylaxis, have occurred; see Monitor ■ Manufactured from human plasma. Risk of transmitting infectious agents (e.g., HIV, hepatitis and, theoretically, Creutzfeldt-Jakob disease) has been greatly reduced by screening, testing, and manufacturing techniques. However, risk of transmission cannot be totally eliminated. Health care professionals should use caution during administration; may have risk of exposure to viral infection. ■ Contains trace human proteins other than factor X. ■ Neutralizing antibodies (inhibitors) to factor X may occur; see Monitor.
Monitor:
Monitor BP and pulse during infusion. If a marked increase in pulse occurs, either reduce rate of infusion or interrupt the infusion. ■ Throughout the infusion, monitor for S/S of a hypersensitivity reaction (e.g., angioedema, chest tightness, chills, fever, headache, hypotension, lethargy, nausea, pruritus, rash, restlessness, tachycardia, urticaria, wheezing). ■ Monitor plasma factor X activity by performing a validated test (e.g., one-stage clotting assay) to confirm that adequate factor X levels have been achieved and maintained. ■ In surgery patients, monitor postinfusion plasma factor X levels before and after surgery to ensure that hemostatic levels are obtained and maintained. ■ Monitor for the development of factor X inhibitors. Perform a Nijmegen-Bethesda inhibitor assay if expected factor X plasma levels are not attained or if bleeding is not controlled with the expected dose of coagulation factor X.
Patient education:
Review manufacturer’s medication guide. ■ Promptly report S/S of a hypersensitivity reaction (e.g., dizziness, hives, itching, rash, tightness of the chest, wheezing). ■ Report a lack of clinical response to therapy; may be a manifestation of an inhibitor. ■ Report symptoms of possibly transmitted viral infections immediately. Symptoms may include anorexia, arthralgias, fatigue, jaundice, low-grade fever, nausea, or vomiting. ■ If traveling, bring an adequate supply of medication based on current treatment regimen.
Maternal/child:
Use during pregnancy or labor and delivery only if clearly needed. ■ Safety for use during breast-feeding not known; consider benefit versus risk. ■ Safety and effectiveness for use in pediatric patients under 12 years of age not established.
Elderly:
Numbers insufficient to determine differences in response between older and younger patients.
Drug/lab interactions
Drug interaction studies have not been performed. ■ Use with caution in patients who are receiving other plasma products that may contain factor X (e.g., fresh frozen plasma, prothrombin complex concentrates [e.g., Kcentra]). ■ Based on its mechanism of action, coagulation factor X is likely to be counteracted by direct and indirect factor Xa inhibitors (e.g., apixaban [Eliquis], edoxaban [Savaysa], rivaroxaban [Xarelto]).
Side effects
The most common side effects observed are back pain, fatigue, infusion site erythema, and infusion site pain. Hypersensitivity reactions have occurred.
Antidote
Keep the physician informed of side effects. Slow or interrupt infusion for a marked increase in pulse rate or a mild hypersensitivity reaction. Discontinue the infusion immediately if a severe hypersensitivity reaction occurs. Treat hypersensitivity as necessary (e.g., antihistamines, epinephrine, corticosteroids). Resuscitate as necessary.
Coagulation factor XIII A-subunit (recombinant)
(ko-ag-yew-LA-shun FAK-ter XIII [re-KOM-be-nant])
Tretten
Antihemorrhagic
pH 8
Usual dose (international units [IU])
Adult and pediatric patients:
35 IU/kg once monthly to achieve a target trough level of factor XIII activity at or above 10% using a validated assay.
Dose adjustments
Consider increasing dose if adequate trough levels are not achieved.
Dilution
Available as a white lyophilized powder in single-use vials. May contain from 2,000 to 3,125 IU. Actual amount is stated on each vial and each carton. Can be reconstituted using the vial adapter and diluent included with the packaging (see prescribing information) or using a needle and syringe with SWFI as a diluent. Bring vial and diluent to RT (not to exceed 25° C (77° F). Withdraw 3.2 mL of SWFI and inject into the powder by aiming the needle and stream of diluent against the side of the vial. To avoid foaming, do not inject the diluent directly on the powder. Gently swirl to dissolve. Do not shake. A clear and colorless solution. After reconstitution, each mL contains 667 to 1,042 IU.
If a large dose requires the use of multiple vials, reconstitute each additional vial with a separate syringe. For a smaller dose that requires less than the full volume in the vial, the reconstituted solution may be further diluted with NS to facilitate measurement and administration; discard remaining product. Must be used within 3 hours.
Filters:
Specific information not available.
Storage:
Refrigerate at 2° to 8° C (36° to 46° F) in original carton to protect from light. Do not freeze. Stable until expiration date on carton. Immediate use of reconstituted solution is preferred but may be refrigerated or kept at RT (not to exceed 25° C [77° F]) for up to 3 hours. Discard after 3 hours.
Compatibility
Manufacturer states, “Do not administer with other infusion solutions.”
Rate of administration
A single dose as an IV injection at a rate not to exceed 1 to 2 mL/min. Do not administer as a drip infusion.
Actions
A human factor XIII-A2 homodimer produced by recombinant DNA technology. Factor XIII is the terminal enzyme in the blood coagulation cascade. When activated by thrombin at the site of vessel wall injury, factor XIII plays an important role in the maintenance of hemostasis through cross-linking of fibrin and other proteins in the fibrin clot. After combining with available factor XIII B-subunits, it has been shown to have the same pharmacodynamics properties in plasma as endogenous factor XIII. Increases the mechanical strength of fibrin clots, retards fibrinolysis, and enhances platelet adhesion to the site of injury. Mean half-life based on baseline (trough) adjusted factor XIII activity was 5.1 days in patients 7 to 58 years of age and 7.1 days in pediatric patients 1 to less than 6 years of age.
Indications and uses
Routine prophylaxis for bleeding in patients with congenital factor XIII A-subunit deficiency.
Limitation of use:
Not for use in patients with congenital factor XIII B-subunit deficiency.
Contraindications
Known hypersensitivity to the active substance or any of its excipients.
Precautions
For IV injection only. Do not administer as a drip infusion. ■ Should be administered under the supervision of a physician experienced in the treatment of rare bleeding disorders. ■ Hypersensitivity reactions (including anaphylaxis) may occur. ■ Thrombotic events may occur. Use caution in patients with an increased risk of thromboembolic complications. These include, but are not limited to, patients with a history of coronary artery disease (CAD), liver disease, DIC, or postoperative immobilization; elderly patients; and neonates. ■ Inhibitory antibodies may occur and should be considered if there is an inadequate response or a reduced therapeutic effect to treatment.
Monitor:
Confirm factor XIII A-subunit deficiency. ■ Evaluate effectiveness of treatment to achieve a target trough level of factor XIII activity at or above 10% using a validated assay at least monthly or more frequently if indicated. Adjust dose as indicated. ■ Monitor for S/S of hypersensitivity (e.g., hypotension, rash, urticaria, tightness of the chest, wheezing). ■ Monitor for S/S of thromboembolic complications (e.g., chest pain, limb or abdominal swelling and/or pain, shortness of breath, loss of sensation or motor power, altered consciousness, vision, or speech). ■ Monitor for S/S of inhibitory antibodies (e.g., expected plasma factor XIII activity levels are not attained, or breakthrough bleeding occurs during prophylactic treatment). Confirm with an assay that measures factor XIII inhibitory antibody concentrations.
Patient education:
Review manufacturer’s medication guide. ■ Promptly report S/S of a hypersensitivity reaction (e.g., light-headedness, rash, urticaria, tightness of the chest, wheezing). ■ Signs of bleeding may be similar to signs of thrombosis and can include new-onset swelling and pain in the limbs or abdomen, new-onset chest pain, shortness of breath, loss of sensation or motor power, or altered consciousness, vision, or speech. Promptly report any of these signs. ■ Report breakthrough bleeding; it may be a symptom of inhibitor formation and require further testing.
Maternal/child:
Category C: safety for use during pregnancy not established. Use only if clearly needed. ■ Use caution during breast-feeding. ■ Clinical studies included small numbers of pediatric patients from neonates to adolescents who were treated with multiple exposures. Side effects were reported more frequently in pediatric patients 6 to less than 18 years of age.
Elderly:
Numbers in clinical studies insufficient to determine if the elderly respond differently from younger subjects.
Drug/lab interactions
Concomitant administration with coagulation factor VIIa may cause thrombosis.
Side effects
Headache, increase in fibrin d-dimer levels, injection site pain, and pain in the extremities were most commonly reported and were reported more frequently in pediatric patients 6 to less than 18 years of age. Hypersensitivity reactions (including anaphylaxis) and thromboembolic events are the most serious side effects. Formation of inhibitory antibodies has occurred.
Antidote
Discontinue drug and notify physician of any major side effect. Treat hypersensitivity reactions with oxygen, epinephrine (Adrenalin), antihistamines (e.g., diphenhydramine [Benadryl]), vasopressors (e.g., dopamine), corticosteroids, albuterol, IV fluids, and ventilation equipment as indicated. Resuscitate as necessary.
Conivaptan hydrochloride
(kon-ih-VAP-tan hy-droh-KLOR-eyed)
Vaprisol
Arginine vasopressin antagonist
pH 3 to 3.8
Usual dose
Loading dose:
20 mg as an IV infusion over 30 minutes. Follow with 20 mg administered as a continuous infusion evenly distributed over 24 hours. May be administered for an additional 1 to 3 days as a continuous infusion of 20 mg/day. The total duration of infusion (after the loading dose) should not exceed 4 days.
Dose adjustments
If the serum sodium does not rise at the desired rate, the dose may be titrated up to 40 mg as a continuous infusion over 24 hours. 40 mg is the maximum daily dose. ■ A reduced dose may be required if the patient experiences an undesirably rapid rate of rise of serum sodium; see Precautions. ■ If hyponatremia persists or recurs (after initial interruption of therapy) and the patient has no evidence of neurologic sequelae, conivaptan may be resumed at a reduced dose; see Monitor. ■ A reduced dose may also be required in patients who develop hypotension or hypovolemia; see Monitor. ■ No dose adjustment indicated in patients with mild hepatic impairment. ■ Reduced dose required in patients with moderate hepatic impairment. Initiate with a loading dose of 10 mg. Follow with a continuous infusion of 10 mg over 24 hours for 2 days to a maximum of 4 days. If sodium is not rising at the desired rate, the conivaptan dose may be titrated up to 20 mg/day. ■ Use in patients with severe renal impairment (CrCl less than 30 mL/min) is not recommended; see Contraindications.
Dilution
Available in a single-use, ready-to-use (RTU) plastic container containing 20 mg conivaptan in 100 mL D5W. If the RTU container is being administered as a 40-mg dose, administer two consecutive 20 mg/100 mL containers over 24 hours.
Storage:
Store in carton at CRT. Protect from light and freezing. Do not remove container from overwrap until ready to use. Overwrap is a moisture and light barrier.
Compatibility
Manufacturer states, “Should not be mixed or administered with lactated Ringer’s or furosemide (Lasix). Should not be combined with any other product in the same intravenous line or bag.”
Rate of administration
Loading dose:
A single dose equally distributed over 30 minutes as an infusion.
Continuous infusion:
Actions
A nonpeptide, dual antagonist of arginine vasopressin (AVP) V1A and V2 receptors. The level of AVP in the blood is critical for the regulation of water and electrolyte balance and is usually elevated in both euvolemic and hypervolemic hyponatremia (in euvolemic hyponatremia there is an increase in total body water, but the sodium content remains the same; in hypervolemic hyponatremia both sodium and water content in the body increase, but the water gain is greater). AVP excess is associated with hyponatremia without edema. The AVP effect is mediated through V2 receptors that help maintain plasma osmolality. Conivaptan blocks V2 receptors in the renal collecting ducts, resulting in aquaresis (excretion of free water). This is generally accompanied by increased net fluid loss, increased urine output, and decreased urine osmolality. Conivaptan is highly protein bound. It is metabolized in the liver by the cytochrome P450 isoenzyme, CYP3A. Its half-life is 5.3 to 8.1 hours, depending on dose. Primarily excreted in the feces and, to a lesser extent, in urine. Crosses the placenta in animals.
Indications and uses
For use in the hospitalized patient to treat euvolemic and hypervolemic hyponatremia. Euvolemic hyponatremia may occur in the syndrome of inappropriate secretion of antidiuretic hormone (SIADH, an inability of the body to excrete dilute urine) or in the setting of certain conditions, including hypothyroidism, adrenal insufficiency, and pulmonary disorders. ■ Not indicated for the treatment of the S/S of heart failure. Raising serum sodium with conivaptan has not been shown to provide a symptomatic benefit.
Contraindications
Patients with hypovolemic hyponatremia. ■ Hypersensitivity to conivaptan or any of its components (propylene glycol, ethanol, lactic acid). ■ Coadministration with potent CYP3A inhibitors, such as clarithromycin, indinavir, itraconazole, ketoconazole, and ritonavir; see Drug Interactions. ■ Premixed solution contains dextrose, which may be contraindicated in patients with a known allergy to corn or corn products. ■ Anuria (no benefit expected).
Precautions
For IV use only. ■ Use only in hospitalized patients. ■ Safety for use in hypovolemic hyponatremic patients with underlying CHF has not been established. Should be used to raise sodium in these patients only after other treatment options have been considered. Incidence of adverse cardiac events may be increased. ■ An overly rapid increase in serum sodium concentration (more than 12 mEq/L/24 hr) may result in serious sequelae. Although not observed in clinical trials, osmotic demyelination syndrome (brain cell dehydration) has been reported following rapid correction of low serum sodium concentration. Osmotic demyelination results in dysarthria, lethargy, affective changes, spastic quadriparesis, seizures, coma, or death. Patients with severe malnutrition, alcoholism, or advanced liver disease may be at increased risk; use slower rates of correction; see Monitor. ■ Reduced doses required in patients with moderate hepatic impairment; see Dose Adjustments. Impact of severe hepatic impairment has not been studied. ■ May cause significant infusion site reaction; see Monitor.
Monitor:
Administer via a large vein. Monitor infusion site and rotate every 24 hours. ■ Monitor serum sodium concentration and neurologic status closely during therapy. If an overly rapid increase in serum sodium concentration occurs (greater than 12 mEq/L/24 hr), administration should be discontinued. If the sodium continues to rise, administration should not be resumed. If hyponatremia persists or recurs (after initial interruption of therapy) and the patient has no evidence of neurologic sequelae, conivaptan may be resumed at a reduced dose. ■ Monitor vital signs, urine output and osmolality, and volume status of patient. Discontinue therapy in patients who develop hypotension or hypovolemia. Once the patient is again euvolemic and is no longer hypotensive, therapy may be resumed at a reduced dose; see Dose Adjustments.
Patient education:
Promptly report any burning at the infusion site or other side effects. ■ Request assistance for ambulation. ■ Review list of allergies and medications with physician or pharmacist.
Maternal/child:
Category C: use during pregnancy only if benefits justify risk to the fetus. Has been shown to cause fetal harm in animals. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients have not been studied.
Elderly:
Response similar to that seen in the general study population.
Drug/lab interactions
A substrate of CYP3A. Coadministration with inhibitors of this enzyme could lead to an increase in conivaptan concentrations, the effect of which is unknown. Concomitant use with potent CYP3A4 inhibitors such as clarithromycin (Biaxin), indinavir (Crixivan), itraconazole (Sporanox), ketoconazole (Nizoral), and ritonavir (Norvir) is contraindicated. ■ A potent inhibitor of CYP3A. May increase plasma concentrations of drugs that are primarily metabolized by this enzyme. Coadministration with amlodipine (Norvasc), midazolam (Versed), and simvastatin (Zocor) resulted in increased concentrations of each of the drugs. Two cases of rhabdomyolysis occurred in patients who were receiving a CYP3A4-metabolized HMG-CoA reductase inhibitor (e.g., simvastatin [Zocor]). Avoid concomitant use with drugs eliminated primarily by CYP3A4-mediated metabolism. Do not initiate subsequent therapy with CYP3A4 substrate drugs until at least 1 week after an infusion of conivaptan. ■ May decrease clearance and increase serum concentration of digoxin. Monitor digoxin levels. ■ Captopril and furosemide (Lasix) do not appear to affect the pharmacokinetics of conivaptan. ■ Does not appear to affect PT/INR when coadministered with warfarin (Coumadin). ■ Does not appear to affect the QT interval.
Side effects
The most common adverse reactions are infusion site reactions (e.g., erythema, pain, phlebitis), fever, headache, hypokalemia, orthostatic hypotension, peripheral edema. Other reactions that occurred in more than 2% of patients include anemia, atrial fibrillation, confusion, constipation, diarrhea, hypertension, hypomagnesemia, hyponatremia, hypotension, insomnia, nausea, pharyngolaryngeal pain, pneumonia, pruritus, pyrexia, ST segment depression on ECG, thirst, urinary tract infection, and vomiting.
Antidote
Notify physician of any side effects. Most will be treated symptomatically. Discontinue therapy if there is an overly rapid increase in serum sodium concentration or if the patient experiences hypotension or hypovolemia. Discontinue therapy permanently if neurologic sequelae are present; see Precautions, Monitor, and Dose Adjustments. Resuscitate as necessary.
Conjugated estrogens 
(KON-jyou-gay-ted ES-troh-jens)
Premarin Intravenous
Hormone (estrogen)
Antihemorrhagic
pH 7.2 to 7.4
Usual dose
25 mg in 1 injection. May be repeated in 6 to 12 hours if indicated.
Dilution
Reconstitute with 5 mL SWFI, directing the flow of diluent gently against the side of the vial. Agitate gently. Do not shake violently. Do not use if discolored or if precipitate is present. Dilution in an IV infusion is not recommended.
Storage:
Refrigerate prior to use. Use immediately after reconstitution.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer lists as incompatible with ascorbic acid, protein hydrolysate, or any solution with an acid pH.
According to the manufacturer, infusion with other agents is not generally recommended. May be given at Y-tube if compatible solutions are infusing (NS, dextrose, and invert sugar solutions). According to one source, it is compatible at the Y-site with heparin, hydrocortisone sodium succinate (Solu-Cortef), and potassium chloride (KCl).
Rate of administration
5 mg or fraction thereof over 1 minute. Must be given direct IV or through IV tubing close to needle site. Infusion solution must be compatible. Too-rapid injection may cause flushing.
Actions
A mixture of conjugated estrogens obtained from natural sources. Administration provides a rapid and temporary increase in estrogen levels. Acts at several points on the clotting cascade, enhancing coagulability of the blood, especially in the capillary beds. Promptly corrects bleeding due to estrogen deficiency. Widely distributed in the body. Metabolized primarily in the liver and excreted in the urine. Secreted in breast milk.
Indications and uses
Treatment of abnormal uterine bleeding caused by hormonal imbalance in the absence of organic pathology. Indicated for short-term use only to provide a rapid and temporary increase in estrogen levels.
Unlabeled uses:
Postcoital contraception.
Contraindications
Known, suspected, or history of breast cancer, active deep venous thrombosis, or pulmonary embolism. ■ A history of or active arterial thromboembolic disease (e.g., stroke, MI). ■ Known or suspected estrogen-dependent neoplasia, pregnancy, undiagnosed abnormal genital bleeding, liver dysfunction or disease. ■ Hypersensitivity to this product or its ingredients (e.g., known anaphylactic reaction and angioedema). ■ Known protein C, protein S, or antithrombin deficiency, or other known thrombophilic disorders. ■ Other specific contraindications for estrogens must be considered.
Precautions
IV therapy with conjugated estrogens is indicated for short-term use. However, warnings and precautions associated with oral therapy should be considered (e.g., changes in vaginal bleeding, headache, hypersensitivity reactions, skin reactions, and many more); see Precautions and prescribing information. ■ Estrogens with or without progestins should not be used for the prevention of cardiovascular disease or dementia. ■ Even though bleeding is controlled, the etiology of the bleeding must be determined and definitive therapy instituted. ■ May cause fluid retention. Use with caution in patients with cardiac or renal dysfunction. ■ Use with caution in asthma, diabetes, endometriosis, epilepsy, hepatic hemangiomas, hepatic or gallbladder disease, hypercalcemia or hypocalcemia, hypercholesterolemia, hypertension, hypertriglyceridemia, hypoparathyroidism, migraines, obesity, porphyria, systemic lupus erythematosus, or tobacco use; may exacerbate condition. ■ May induce or exacerbate symptoms of angioedema, particularly in women with hereditary angioedema. ■ Retinal vascular thrombosis has been reported in patients receiving estrogen therapy. Discontinue therapy and obtain ophthalmologic exam if visual disturbances occur. ■ Patients dependent on thyroid hormone replacement therapy (e.g., levothyroxine [Synthroid]) who are also receiving estrogen may require dose adjustment of thyroid replacement medication. ■ Estrogens may increase the risk of deep venous thrombosis, MI, pulmonary embolism, stroke, breast cancer, endometrial cancer, and dementia. ■ Follow immediately with oral estrogens as recommended for dysfunctional uterine bleeding.
Monitor:
Monitor VS; may cause a temporary BP elevation.
Patient education:
Review health history and disease states with physician before beginning treatment with conjugated estrogens. ■ Review possible side effects with physician and report any side effects promptly.
Maternal/child:
Category X: avoid pregnancy. ■ Use caution during breast-feeding. Detectable amounts have been found in breast milk and may decrease quantity and quality of milk. ■ Safety for use in pediatric patients not established. Extended use may accelerate epiphyseal closure, which could result in short adult stature.
Elderly:
Differences in response compared to younger adults not identified.
Drug/lab interactions
May decrease effects of oral antidiabetics. ■ Barbiturates (e.g., phenobarbital), carbamazepine (Tegretol), phenytoin (Dilantin), rifampin (Rifadin), and St. John’s wort increase metabolism and decrease serum levels and effects. ■ Plasma concentrations may be increased with coadministration of clarithromycin (Biaxin), erythromycin, itraconazole (Sporanox), ketoconazole (Nizoral), ritonavir (Norvir), and grapefruit juice. ■ May decrease metabolism and increase serum levels of cyclosporine. May increase risk of cyclosporine toxicity. ■ Increased risk of hepatotoxicity with other hepatotoxic agents (e.g., dantrolene [Dantrium]). ■ May increase blood glucose levels and serum lipids. ■ May reduce response to metyrapone test. ■ May alter numerous coagulation tests, glucose tolerance, and thyroid and other protein-binding tests.
Side effects
Rare when used as directed; flushing, nausea, vomiting. IV therapy with conjugated estrogens is indicated for short-term use. However, the numerous adverse reactions associated with oral therapy should be considered (e.g., changes in vaginal bleeding, headache, hypersensitivity reactions, skin reactions, and many more); see Precautions and prescribing information.
Post-marketing:
Anaphylaxis within minutes to hours of infusion; angioedema involving the face, tongue, larynx, hands, and feet.
Antidote
No toxicity has been reported throughout years of clinical use. Discontinue if jaundice occurs. Discontinue immediately if DVT, MI, PE, stroke, or venous thromboembolism occurs.
Cosyntropin
(koh-SIN-troh-pin)
Cortrosyn
Diagnostic agent (adrenocorticotropic)
pH 5.5 to 7.5
Usual dose
250 mcg (0.25 mg). Up to 750 mcg (0.75 mg) has been used.
Pediatric dose
Pediatric patients over 2 years of age:
May use adult dose, but 125 mcg (0.125 mg) is usually adequate.
Pediatric patients 2 years of age or less:
125 mcg (0.125 mg). Usually given IM.
Dilution
Available as a lyophilized powder or as a solution. Reconstitute powder with 1 mL NS. For IV use, both formulations should be diluted with 2 to 5 mL of NS. May be given directly IV after this initial dilution or further diluted in D5W or NS and given as an infusion (250 mcg in 250 mL equals 1 mcg/mL).
Storage:
Lyophilized powder: Store at CRT. Liquid formulation: Store at 2° to 8° C (36° to 46° F); protect from light and freezing. Discard any unused drug.
Compatibility
Manufacturer states, “Should not be added to blood or plasma; may be inactivated by enzymes.” Consider specific use.
Rate of administration
IV injection:
A single dose over 2 minutes.
Infusion:
A single dose at a rate of approximately 40 mcg/hr over 6 hours.
Actions
A synthetic form of adrenocorticotropic hormone (ACTH). Stimulates the adrenal cortex to secrete adrenocortical hormone. Does not increase cortisol secretion in patients with primary adrenocortical insufficiency. Peak plasma cortisol levels occur in 1 to 2 hours depending on formulation used.
Indications and uses
Diagnostic aid for adrenocortical insufficiency.
Contraindications
Hypersensitivity to cosyntropin.
Precautions
The liquid formulation of cosyntropin is for IV use only. Cortrosyn may be used IV or IM. ■ Preferable to ACTH because it is less likely to cause hypersensitivity reactions; however, hypersensitivity reactions have occurred. Administer in a facility equipped to monitor the patient and respond to any medical emergency. ■ May be used in patients who have had a hypersensitivity reaction to ACTH.
Monitor:
Continuous observation for at least the first 30 minutes is mandatory. Observe frequently thereafter. ■ Check BP frequently; may cause elevated BP and salt and water retention. ■ Monitor correct collection of specimens; see prescribing information for specific details. ■ See Drug/Lab Interactions.
Patient education:
Promptly report S/S of a hypersensitivity reaction (e.g., hives, rash, shortness of breath or troubled breathing, swelling of eyelids, lips, or face).
Maternal/child:
Category C: use during pregnancy only if benefits outweigh risks. ■ Use with caution during breast-feeding.
Drug/lab interactions
Plasma cortisol may be falsely elevated with cortisone, hydrocortisone, or spironolactone. Patients receiving cortisone, hydrocortisone, or spironolactone should omit their pretest doses on the day of testing. ■ Abnormally high basal plasma cortisol levels may also occur in patients taking inadvertent doses of cortisone or hydrocortisone on the test day and in women taking drugs that contain estrogen. ■ Many drug reactions are possible with corticosteroids, but usually not a concern with specific diagnostic use. ■ May accentuate electrolyte loss associated with diuretic therapy.
Side effects
Bradycardia, hypertension, peripheral edema, rash, and tachycardia have been reported. Hypersensitivity reactions, including anaphylaxis (rare), have occurred.
Antidote
Notify the physician of any side effect. Keep epinephrine and diphenhydramine available to treat anaphylaxis. Resuscitate as necessary.
Cyclophosphamide
(sye-kloh-FOS-fah-myd)
Lyophilized Cytoxan,  Procytox
Procytox
Antineoplastic (alkylating agent/ nitrogen mustard)
pH 3 to 7.5
Usual dose
Although effective alone in susceptible malignancies, cyclophosphamide is more frequently used concurrently or sequentially with other antineoplastic agents. Doses may be expressed in mg/kg or mg/M2. During or immediately after administration, adequate amounts of fluid should be ingested or infused to force diuresis and thus reduce the risk of urinary toxicity. Cyclophosphamide should be administered in the morning; see Monitor.
Malignant diseases (adult and pediatric patients):
As a single agent: The initial dose may be 40 to 50 mg/kg of body weight, usually given in divided doses over 2 to 5 days. Alternate dosing schedules are 3 to 5 mg/kg twice weekly or 10 to 15 mg/kg every 7 to 10 days. Higher doses have been used with some protocols based on the condition being treated. Adequate hydration is indicated, and the use of mesna can be considered to attenuate or reduce the incidence of hemorrhagic cystitis. Combination protocols: Numerous combination therapies are in use. Has been used with bleomycin, bortezomib, carboplatin, cisplatin, dacarbazine, dexamethasone, docetaxel, doxorubicin, epirubicin, etoposide, fludarabine, fluorouracil, methotrexate, prednisone, procarbazine, rituximab, topotecan, vinblastine, and vincristine. See literature for various regimens.
Adjuvant treatment of operable node-positive breast cancer:
Treatment protocol includes cyclophosphamide, doxorubicin, and docetaxel. Administer cyclophosphamide 500 mg/M2 and doxorubicin 50 mg/M2. One hour later, give docetaxel 75 mg/M2. Repeat every 3 weeks for 6 cycles. See docetaxel (Taxotere) and doxorubicin (Adriamycin) monographs.
Pediatric dose
Malignant diseases:
See Usual Dose.
Dose adjustments
Dosages must be adjusted based on antitumor activity and/or leukopenia. Total leukocyte count is a good, objective guide for regulating dosage. Withhold therapy in patients with neutrophils less than or equal to 1,500/mm3 and platelets less than 50,000/mm3. ■ When used as a component of a multidrug regimen, it may be necessary to reduce the dose of cyclophosphamide as well as the doses of the other drugs. ■ Dosing should be cautious in the elderly. Lower-end initial doses may be indicated. Consider decrease in cardiac, hepatic, and renal function; concomitant disease; or other drug therapy; see Elderly.
Dilution
Specific techniques required; see precautions.
Contains no preservatives; aseptic technique imperative. Each 100 mg must be diluted with 5 mL of NS or SWFI; yields 20 mg/mL (a 2% solution). Shake solution gently and allow to stand until clear. A 2% solution reconstituted with NS may be injected directly or further diluted to a minimum concentration of 0.2% (2 mg/mL) for infusion. A 2% solution reconstituted with SWFI is hypotonic and should not be injected directly. It must be further diluted to a minimum concentration of 0.2% (2 mg/mL). D5W, 1/2NS, and D5NS are compatible diluents. Do not use heat to facilitate dilution. See Monitor.
Filters:
May be filtered through available micron sizes of cellulose ester membrane filters.
Storage:
Store unopened vials at or below 25° C (77° F). Cyclophosphamide that is reconstituted in SWFI must be used immediately. Do not store. If reconstituted in NS or further diluted, it must be used within 24 hours when stored at RT. Solutions reconstituted in NS or further diluted in 1/2NS are stable up to 6 days if refrigerated. Solutions further diluted in D5W or D5NS are stable for 36 hours if refrigerated. Do not use cyclophosphamide vials if there are signs of melting (clear or yellowish viscous liquid or droplets).
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Bleomycin (Blenoxane), cisplatin, fluorouracil (5-FU), mesna (Mesnex), methotrexate, mitoxantrone (Novantrone), ondansetron (Zofran).
Y-site:
Allopurinol (Aloprim), amifostine (Ethyol), amikacin, ampicillin, anidulafungin (Eraxis), aztreonam (Azactam), bleomycin (Blenoxane), cefazolin (Ancef), cefotaxime (Claforan), cefoxitin (Mefoxin), cefuroxime (Zinacef), chloramphenicol (Chloromycetin), cisplatin, cladribine (Leustatin), clindamycin (Cleocin), doripenem (Doribax), doxorubicin (Adriamycin), doxorubicin liposomal (Doxil), doxycycline, droperidol (Inapsine), erythromycin (Erythrocin), etoposide phosphate (Etopophos), filgrastim (Neupogen), fludarabine (Fludara), fluorouracil (5-FU), furosemide (Lasix), gallium nitrate (Ganite), gemcitabine (Gemzar), gentamicin, granisetron (Kytril), heparin, idarubicin (Idamycin), leucovorin calcium, linezolid (Zyvox), melphalan (Alkeran), methotrexate, metoclopramide (Reglan), metronidazole (Flagyl IV), mitomycin (Mutamycin), nafcillin (Nallpen), ondansetron (Zofran), oxacillin (Bactocill), oxaliplatin (Eloxatin), paclitaxel (Taxol), palonosetron (Aloxi), pemetrexed (Alimta), penicillin G potassium, piperacillin/tazobactam (Zosyn), propofol (Diprivan), sargramostim (Leukine), sodium bicarbonate, sulfamethoxazole/trimethoprim, teniposide (Vumon), thiotepa, ticarcillin/clavulanate (Timentin), tobramycin, topotecan (Hycamtin), vancomycin, vinblastine, vincristine, vinorelbine (Navelbine).
Rate of administration
Rate may vary depending on protocol. May be given by IV push or as an intermittent or continuous infusion. Inject or infuse very slowly to reduce the likelihood of adverse reactions that may be administration rate–dependent (e.g., facial swelling, headache, nasal congestion, scalp burning). Duration of infusion should be appropriate for the volume to be infused.
Actions
An alkylating agent of the nitrogen mustard group with antitumor activity; cell cycle phase nonspecific, but most effective in S phase. Cyclophosphamide is a prodrug that is activated by hepatic microsomal enzymes. It is thought to prevent cell division by cross-linking DNA strands and preventing DNA synthesis. Elimination half-life is 3 to 12 hours. Metabolized in the liver, it or its metabolites are excreted in the urine. Secreted in breast milk.
Indications and uses
Although effective alone in susceptible malignancies, cyclophosphamide is more frequently used concurrently or sequentially with other antineoplastic agents. Cyclophosphamide has been used in the treatment of the following malignancies: malignant lymphomas (e.g., Hodgkin’s disease, lymphocytic lymphoma, mixed-cell–type lymphomas, histiocytic lymphoma, Burkitt’s lymphoma), multiple myeloma, leukemias (e.g., chronic lymphocytic leukemia, chronic granulocytic leukemia, acute myelogenous and monocytic leukemia, acute lymphoblastic [stem cell] leukemia in children), mycosis fungoides, neuroblastoma, adenocarcinoma of the ovary, retinoblastoma, and carcinoma of the breast. ■ Adjuvant treatment of operable node-positive breast cancer in combination with doxorubicin and docetaxel. ■ Used orally to treat many other indications, including biopsy-proven nephrotic syndrome, in pediatric patients when disease fails to respond to primary therapy or when primary therapy causes intolerable side effects.
Limitations of use:
Safety and effectiveness for the treatment of nephrotic syndrome in adults or of other renal disease have not been established.
Unlabeled uses:
Ewing’s sarcoma, granulomatosis with polyangiitis (GPA, Wegener’s granulomatosis), lupus nephritis, non-Hodgkin’s lymphoma, severe rheumatologic conditions. ■ Transplant conditioning and many others; see literature.
Contraindications
Hypersensitivity to cyclophosphamide, any of its metabolites, or to other components of the product. ■ Urinary outflow obstruction.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of the physician specialist. ■ Myelosuppression (leukopenia, neutropenia, thrombocytopenia, and anemia), bone marrow failure, and severe immunosuppression may lead to serious and sometimes fatal infections. Sepsis and septic shock can occur. Latent infections (e.g., tuberculosis, viral hepatitis) can be reactivated. ■ Hemorrhagic cystitis, pyelitis, ureteritis, and hematuria have been reported. Urotoxicity can be fatal. ■ Exclude or correct any urinary tract obstructions before starting treatment; see Contraindications. ■ Use with caution, if at all, in patients with active urinary tract infections. ■ Myocarditis, myopericarditis, pericardial effusions including cardiac tamponade, and congestive heart failure have been reported and may be fatal. Supraventricular and ventricular arrhythmias (including severe QT prolongation) have been reported. Risk of cardiotoxicity may be increased with high doses, in the elderly, in patients with pre-existing cardiac disease, and in patients with previous radiation treatment to the cardiac region and/or previous or concomitant treatment with other cardiotoxic drugs (e.g., bleomycin, doxorubicin). ■ Pneumonitis, pulmonary fibrosis, pulmonary veno-occlusive disease, and other forms of pulmonary toxicity leading to respiratory failure have been reported during and after treatment with cyclophosphamide. Late-onset pneumonitis (greater than 6 months after the start of therapy) appears to be associated with increased mortality. Pneumonitis may develop years after treatment. ■ Development of secondary malignancies has been reported in patients treated with cyclophosphamide used alone or in association with other antineoplastic agents. May develop several years after treatment has been discontinued. ■ Risk of bladder cancer may be reduced by prevention of hemorrhagic cystitis. ■ Veno-occlusive liver disease (VOD) has been reported. Fatalities have occurred. A cytoreductive regimen in preparation for bone marrow transplantation that consists of cyclophosphamide in combination with whole-body irradiation, busulfan, or other agents has been identified as a major risk factor. VOD has also been reported to develop gradually in patients receiving long-term, low-dose, immunosuppressive doses of cyclophosphamide; in patients with pre-existing hepatic impairment; in patients who have received abdominal radiation; and in patients with low performance status. ■ Hyponatremia associated with increased total body water, acute water intoxication, and a syndrome that resembles syndrome of inappropriate secretion of antidiuretic hormone (SIADH) has been reported. ■ Use caution in patients with impaired renal function. ■ Use with caution in patients with impaired hepatic function. Reduced conversion to active metabolite may cause decreased efficacy. ■ May interfere with normal wound healing. ■ Do not administer any live virus vaccine to patients receiving antineoplastic drugs. ■ Anaphylaxis resulting in death has been reported.
Monitor:
Monitor CBC and platelets and SCr frequently; see Dose Adjustments. ■ Check urinary sediment regularly for presence of erythrocytes and other signs of urotoxicity and/or nephrotoxicity. ■ Aggressive hydration with forced diuresis and frequent bladder emptying can reduce the frequency and severity of bladder toxicity. Mesna has been used to prevent severe bladder toxicity. ■ Monitor patients with severe renal impairment (CrCl 10 to 24 mL/min) for signs and symptoms of toxicity. ■ Observe continuously for infection. Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. Antimycotics and/or antivirals may also be indicated. ■ Monitor patients with risk factors for cardiotoxicity. ■ Monitor patients for signs and symptoms of pulmonary toxicity. ■ Use antiemetics for patient comfort. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood).
Patient education:
Nonhormonal birth control recommended. Female patients should use highly effective contraception during and for up to 1 year after completion of treatment. Male patients with a female partner who is or may become pregnant should use a condom during and for at least 4 months after treatment. ■ Male and female reproductive function and fertility may be impaired. ■ Interferes with oogenesis and spermatogenesis. May cause sterility in both sexes. ■ Increase fluid intake and void frequently. ■ Promptly report any signs or symptoms of infection, any urinary symptoms, new or worsening shortness of breath, cough, swelling of the ankles/legs, palpitations, weight gain, dizziness, loss of consciousness, or other side effects. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: may produce fetal harm. ■ Discontinue breast-feeding. ■ Safety profile in pediatric patients similar to that of adults. ■ See Patient Education.
Elderly:
Consider age-related organ impairment. Dose selection should be cautious; see Dose Adjustments. Toxicity may be increased. Monitoring of renal function is suggested.
Drug/lab interactions
Severe myelosuppression may be expected in patients pretreated with and/or receiving concomitant chemotherapy and/or radiation therapy. ■ Concomitant use of protease inhibitors (e.g., atazanavir [Reyataz], indinavir [Crixivan], nelfinavir [Viracept], ritonavir [Norvir], saquinavir [Invirase]) may increase the concentration of cytotoxic metabolites. Use of protease inhibitor–based regimens was found to be associated with a higher incidence of infections and neutropenia in patients receiving cyclophosphamide, doxorubicin, and etoposide (CDE) than in patients receiving a nonnucleoside reverse transcriptase inhibitor–based regimen (e.g., delavirdine [Rescriptor], nevirapine [Viramune]). ■ Increased hematotoxicity and/or immunosuppression may result from the combined effect of cyclophosphamide and ACE inhibitors (e.g., enalapril [Vasotec], lisinopril [Prinivil, Zestril]), allopurinol (Aloprim), natalizumab (Tysabri), paclitaxel (Taxol), thiazide diuretics (e.g., hydrochlorothiazide), and zidovudine (AZT, Retrovir). ■ Cardiotoxicity may result from a combined effect of cyclophosphamide and anthracyclines (e.g., doxorubicin [Adriamycin], liposomal doxorubicin [Doxil], epirubicin [Ellence]), cytarabine (ARA-C), pentostatin (Nipent), radiation therapy of the cardiac region, and trastuzumab (Herceptin). ■ Increased pulmonary toxicity may result from a combined effect of cyclophosphamide and amiodarone (Nexterone), filgrastim (Neupogen), and sargramostim (Leukine). ■ Increased nephrotoxicity may result from the combined effect of cyclophosphamide and amphotericin B and indomethacin. ■ Risk of hepatotoxicity is increased when administered with azathioprine. ■ Increased incidence of hepatic VOD and mucositis when administered with busulfan (Busulfex, Myleran). ■ Increased incidence of mucositis when administered with protease inhibitors (e.g., atazanavir [Reyataz], indinavir [Crixivan], nelfinavir [Viracept], ritonavir [Norvir], saquinavir [Invirase]). ■ Increased risk of hemorrhagic cystitis may result from a combined effect of cyclophosphamide and past or concomitant radiation treatment. ■ Higher incidence of noncutaneous malignant solid tumors in patients with Wegener’s granulomatosis when administered with etanercept (Enbrel). ■ Acute encephalopathy has been reported in patients receiving metronidazole (Flagyl) and cyclophosphamide. ■ Concomitant use with tamoxifen (Soltamox) may increase the risk of thromboembolic complications. ■ May increase or decrease activity of anticoagulants (e.g., warfarin [Coumadin]); monitor INR and adjust dose as indicated. ■ May reduce serum digoxin (Lanoxin) and cyclosporine (Sandimmune, Neoral) levels. ■ May prolong neuromuscular blockade and prolonged respiratory depression caused by succinylcholine. These effects are dose dependent and may occur up to several days after cyclophosphamide is discontinued. Alert the anesthesiologist. ■ May decrease effectiveness of oral quinolone antibiotics (e.g., ciprofloxacin [Cipro], levofloxacin [Levaquin]). ■ Capable of many other interactions.
Side effects
The most commonly reported side effects are alopecia (regrowth may be slightly darker), diarrhea, febrile neutropenia, fever, nausea, neutropenia, and vomiting. Other side effects include abdominal pain, amenorrhea, anorexia, gonadal suppression, impaired wound healing, leukopenia (see Precautions), malaise, mucosal ulcerations, darkening of skin and fingernails, susceptibility to infection, including opportunistic infections.
Major:
Anaphylaxis, bone marrow suppression (anemia, leukopenia, thrombocytopenia) or failure, hemorrhagic cystitis or ureteritis (reversible), infection, pneumonitis, pulmonary fibrosis, rash, renal tubular necrosis (reversible), secondary neoplasia, SIADH, urinary tract and renal toxicity (e.g., bladder fibrosis, hemorrhagic cystitis, ureteritis), veno-occlusive disease.
Post-marketing:
Numerous side effects have been identified from post-marketing surveillance. These include acute respiratory distress syndrome, arthralgia, ascites, asthenia, bronchospasm, cardiotoxicity (e.g., arrhythmias, cardiac arrest, cardiac failure, cardiogenic shock, cardiomyopathy, myocardial infarction, myocarditis, pericarditis), chest pain, chills, cholestasis, colitis, convulsions, disseminated intravascular coagulation, dizziness, dyspnea, edema, encephalopathy, fatigue, fever, fluid retention, headache, hemolytic-uremic syndrome, hepatic encephalopathy, hepatitis, hepatotoxicity with hepatic failure, hyperglycemia, hypertension, hypoglycemia, hyponatremia, hypotension, increased liver function tests, infusion site reactions, interstitial pneumonitis, malaise, muscle spasms, myalgia, pancreatitis, paresthesia, peripheral neuropathy, pruritus, pulmonary edema, radiation recall dermatitis, reversible posterior leukoencephalopathy syndrome, rhabdomyolysis, Stevens-Johnson syndrome, stomatitis, toxic epidermal necrolysis, visual impairment, and many others.
Antidote
No specific antidote available. Minor side effects will be treated symptomatically if necessary. Discontinue the drug in cases of severe hemorrhagic cystitis. Urotoxicity may require interruption of treatment or cystectomy. Mesna has been used to decrease the incidence of cystitis. Interrupt therapy, reduce dose, or discontinue in patients who have or who develop potentially serious infections. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. Leukocyte and thrombocyte nadirs are usually reached in the first or second week of treatment. Peripheral blood cell counts are expected to normalize after approximately 20 days. Supportive therapy as indicated will help sustain the patient in toxicity. Cyclophosphamide and its metabolites are dialyzable.
Cyclosporine 
(sye-kloh-SPOR-een)
Sandimmune
Immunosuppressant
Usual dose
5 to 6 mg/kg of body weight as a single dose 4 to 12 hours before transplantation. Repeat once each day until oral dosage form can be tolerated. Individualized adjustment is imperative and may be required on a daily basis. Administered at 1/3 of the oral dose in patients temporarily unable to take oral cyclosporine. Administered in conjunction with adrenal corticosteroids; different regimens used; see prescribing information.
Pediatric dose
Same as adult dose; however, higher doses may be required. See Maternal/Child.
Dose adjustments
Reduced dose may be required in impaired renal function and in patients with severe hepatic impairment. ■ Higher doses may be required in pediatric patients. ■ Lower-end doses may be indicated in the elderly. Consider impaired organ function and concomitant disease or other drug therapy. ■ Cyclosporine (Sandimmune) is not bioequivalent to Neoral (a brand of cyclosporine oral solution). Conversion from Neoral dosing to Sandimmune may result in lower cyclosporine blood concentrations. Blood concentration monitoring is indicated to avoid potential underdosing. ■ See Monitor and Drug/Lab Interactions.
Dilution
Each 50 mg should be diluted immediately before use with 20 to 100 mL of NS or D5W and given as an infusion. May leach phthalate from polyvinylchloride containers; use diluents in glass infusion bottles. Dilute immediately before use and discard unused portion.
Filters:
Filtered through a 0.45-micron polyprolene filter during manufacturing. Has a high ethanol content, which the filter must accommodate. A large-bore needle filter may be used when withdrawing cyclosporine from an ampule. Adsorption should be negligible, but if there is concern, draw diluent through the same filter. In-line filtering is acceptable. Manufacturer indicates that cyclosporine molecules are small enough to pass through an in-line filter as small as 0.22 microns. Loss of potency is not expected. Another source used 0.22- and 0.45-micron filters and indicates an initial loss of potency that recovered to full concentration.
Storage:
Before use, store ampules at CRT. Protect from light. Discard diluted solution after 24 hours.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Leaches out plasticizers, including DEHP from PVC infusion bags and IV tubing; use of non-PVC containers and IV tubing recommended.
One source suggests the following compatibilities:
Additive:
Ciprofloxacin (Cipro IV), fat emulsion IV.
Y-site:
Anidulafungin (Eraxis), caspofungin (Cancidas), ceftaroline (Teflaro), doripenem (Doribax), linezolid (Zyvox), meropenem (Merrem IV), micafungin (Mycamine), propofol (Diprivan), sargramostim (Leukine), telavancin (Vibativ).
Rate of administration
A single dose properly diluted as a slow IV infusion equally distributed over 2 to 6 hours.
Actions
A potent immunosuppressive agent. Interferes with IL-2 production and blocks T-cell proliferative signals during early T-cell activation. Prolongs survival of kidney, liver, and heart allogeneic transplants in the human. Measured by specific or nonspecific assays. Extensively metabolized by the cytochrome P450 hepatic enzyme system. Half-life is 19 hours (range 10 to 27 hours). Primarily excreted in bile and to a small extent in urine. Crosses the placental barrier. Secreted in breast milk.
Indications and uses
Prophylaxis of organ rejection in kidney, liver, and heart allogeneic transplants in conjunction with adrenocortical steroids. ■ Treatment of chronic rejection in patients previously treated with other immunosuppressive agents. Reserve parenteral formulation for when oral administration not feasible.
Unlabeled uses:
Decrease frequency of pancreatic or corneal allograft rejection. ■ Prevention of acute graft-versus-host disease. ■ Crohn’s disease.
Contraindications
Hypersensitivity to cyclosporine or to Cremophor EL (polyoxyethylated castor oil).
Precautions
Anaphylactic reactions have been reported with the IV formulation. These reactions may be related to the IV vehicle Cremophor EL; patients who experienced these reactions have subsequently been treated with the oral formulation of cyclosporine without incident. Because of the risk of anaphylaxis, IV cyclosporine should be reserved for patients who are unable to take oral therapy. ■ Usually administered in the hospital by or under the direction of a physician experienced in immunosuppressive therapy and management of organ transplant patients. ■ Adequate laboratory and supportive medical resources must be available. ■ All formulations may be given concomitantly with adrenocortical steroids. Manufacturer has a Black Box Warning. Do not administer cyclosporine with any other immunosuppressive agent except adrenocortical steroids. ■ Can cause hepatotoxicity and nephrotoxicity; see Monitor. In impaired renal function, if rejection is severe, try other immunosuppressive therapy or allow rejection and removal of the kidney rather than increase dose of cyclosporine. ■ May cause lymphomas and other malignancies, particularly those of the skin. Increased risk of developing a malignancy appears to be related to the intensity and duration of immunosuppression. Some malignancies may be fatal. ■ Patients receiving immunosuppressive therapies, including cyclosporine and cyclosporine-containing regimens, are at increased risk for infections (viral, bacterial, fungal, protozoal). Both generalized and localized infections can occur. Opportunistic infections include polyomavirus infections (e.g., JC virus–associated progressive multifocal leukoencephalopathy [PML]; polyoma virus–associated nephropathy [PVAN], especially due to BK virus infection). Pre-existing infections may be aggravated. Latent infections may be reactivated. Fatal outcomes have been reported. ■ Encephalopathy has been reported, including posterior reversible encephalopathy syndrome (PRES). May be manifest as impaired consciousness, convulsions, visual disturbances (including blindness), loss of motor function, movement disorders, and psychiatric disturbances. Predisposing factors may include hypertension, hypomagnesemia, hypocholesterolemia, high-dose corticosteroids, high cyclosporine blood levels, and graft-versus-host disease. Patients receiving liver transplants may be more susceptible to encephalopathy than patients receiving kidney transplants. Reversal of encephalopathy has occurred after discontinuation or dose reduction of cyclosporine. ■ Convulsions have been reported, particularly in patients receiving concomitant therapy with high-dose methylprednisolone. ■ Significant hyperkalemia (sometimes associated with hyperchloremic acidosis) and hyperuricemia have been reported. ■ A syndrome of thrombocytopenia and microangiopathic hemolytic anemia that may result in graft failure has been reported. ■ Optic disc edema, including papilledema, with possible visual impairment secondary to benign intracranial hypertension has been reported. ■ See Drug/Lab Interactions.
Monitor:
Observe for S/S of an anaphylactic reaction (e.g., blood pressure changes; bronchospasm; dyspnea; edema of face, tongue, or throat; itching; rash; tachycardia; wheezing). Monitor continuously for the first 30 minutes of the infusion and frequently thereafter. ■ Can cause hepatotoxicity and nephrotoxicity. Monitor BUN, SCr, serum bilirubin, and liver enzymes frequently. Timing and amount of rise in BUN and creatinine and degree of nephrotoxicity or hepatotoxicity distinguish between need for dose reduction or symptoms of organ rejection. ■ May be difficult to distinguish between nephrotoxicity and rejection. Up to 20% of patients may have simultaneous nephrotoxicity and rejection. See package insert for a chart discussing differential diagnoses for each. ■ Monitor cyclosporine blood levels. Measured by specific or nonspecific assay. 24-hour specific trough values of 100 to 200 ng/mL of whole blood or 24-hour nonspecific trough values of 250 to 800 ng/mL of whole blood minimize side effects and rejection events. Nonspecific assays trough values are higher because they include metabolites. Plasma levels may range from 1/2 to 1/5 of whole blood levels. Consistent use of one assay is recommended. Confirm assay method to evaluate appropriately. ■ Observe constantly for signs of infection (fever, sore throat, tiredness) or unusual bleeding or bruising. ■ Prophylactic antibiotics may be indicated pending results of C/S. ■ Monitor for development of PVAN and BK virus–associated nephropathy (e.g., deteriorating renal function and renal graft loss). Dose reduction may be indicated. ■ Monitor for development of PML (e.g., apathy, ataxia, cognitive deficiencies, confusion, hemiparesis). If symptoms appear, consultation with a neurologist may be indicated. ■ Monitor BP. Hypertension is a common side effect. Initiation or modification of antihypertensive therapy may be indicated; do not use potassium-sparing diuretics (e.g., spironolactone [Aldactone]); may increase risk of hyperkalemia. ■ Monitor for S/S of encephalopathy and/or PRES. ■ See Precautions and Drug/Lab Interactions.
Patient education:
Use nonhormonal birth control. Do not use oral contraceptives. See Appendix D, p. 1333. ■ Do not make any changes in formulation (e.g., IV, capsules, oral solution) without physician direction; products are not equivalent. May require dose adjustment. ■ Review side effects with a health care professional and report all side effects promptly. ■ Capable of multiple drug-drug interactions; obtain physician approval before adding or stopping medications. ■ Compliance with frequent laboratory tests is imperative.
Maternal/child:
Category C: safety for use in pregnancy not established. Should not be used unless benefit to the mother justifies potential risk to the fetus. Use in men and women capable of conception not established. Reported outcomes of pregnancies in women who received cyclosporine are difficult to evaluate. It is not possible to separate the effects of cyclosporine from the effects of other medications, underlying maternal disorders, or other aspects of the transplantation process. Negative outcomes included prematurity, low birth weight, fetal loss, and various malformations. ■ Discontinue breast-feeding. ■ Safety for use in pediatric patients not established but has been used in patients as young as 6 months. Accidental parenteral overdose in premature neonates has caused serious symptoms of intoxication.
Elderly:
Dose selection should be cautious; see Dose Adjustments. ■ Differences in response compared to younger adults not identified.
Drug/lab interactions
Interactions are numerous and potentially life threatening. Review of drug profile by pharmacist imperative. ■ Risk of nephrotoxicity increased when given with other drugs that may potentiate renal dysfunction. Use extreme caution and monitor renal function closely. If impairment of renal function is significant, either reduce the dose of cyclosporine and/or the coadministered drug, or consider an alternative treatment. Manufacturer lists antibiotics (e.g., ciprofloxacin [Cipro], gentamicin, tobramycin, sulfamethoxazole/trimethoprim, vancomycin), antifungals (e.g., amphotericin B, ketoconazole [Nizoral]), anti-inflammatory drugs (e.g., azapropazon [Rheumox], colchicine, diclofenac [Voltaren, Cataflam], naproxen [Aleve, Naprosyn], sulindac [Clinoril]), H2 antagonists (e.g., cimetidine [Tagamet], ranitidine [Zantac]), immunosuppressives (e.g., tacrolimus [Prograf]), antineoplastics (e.g., melphalan [Alkeran], methotrexate), and fibric acid derivatives (e.g., fenofibrate [Tricor]). Other sources list acyclovir (Zovirax), foscarnet (Foscavir), selected quinolones (e.g., norfloxacin [Noroxin]), and numerous other nephrotoxic drugs. ■ Concurrent administration with colchicine may cause cyclosporine toxicity (e.g., GI, hepatic, renal, and neuromuscular toxicity). Cyclosporine may decrease the clearance and increase the toxic effects of colchicine (e.g., myopathy, neuropathy), especially in patients with renal impairment. With concurrent use, close clinical observation is required. Reduce colchicine dose or discontinue as indicated. ■ May increase diclofenac (Voltaren) serum levels with concomitant administration; initiate diclofenac dose at the lower end of the therapeutic range. ■ Cyclosporine is extensively metabolized by CYP3A4 and is a substrate of the multidrug efflux transporter P-glycoprotein. Drugs that inhibit or induce CYP3A4, P-glycoprotein transporter, or organic anion transporter proteins will result in an alteration of cyclosporine concentrations. Toxicity or allograft rejection may occur. Compounds that decrease cyclosporine absorption, such as orlistat (Alli), should be avoided. ■ Cyclosporine plasma levels may be increased with concurrent use of protease inhibitors (e.g., boceprevir [Victrelis], indinavir [Crixivan], nelfinavir [Viracept], ritonavir [Norvir], saquinavir [Invirase], telaprevir [Incivek]), which are metabolized by cytochrome P450 3A; use caution. ■ Drugs that inhibit the cytochrome P450 system may decrease the metabolism of cyclosporine and increase its serum concentrations. Manufacturer lists allopurinol (Aloprim), amiodarone (Nexterone), antibiotics (e.g., azithromycin [Zithromax], clarithromycin, erythromycin, quinupristin/dalfopristin [Synercid]), antifungals (e.g., fluconazole [Diflucan], itraconazole [Sporanox], ketoconazole [Nizoral], voriconazole [VFEND]), bromocriptine (Parlodel), calcium channel blockers (e.g., diltiazem [Cardizem], nicardipine [Cardene], verapamil), danazol (Danocrine), glucocorticoids (e.g., methylprednisolone [Solu-Medrol]), imatinib (Gleevec), metoclopramide (Reglan), nefazodone, and oral contraceptives. Monitor blood levels with concurrent use to avoid cyclosporine toxicity. ■ Drugs that decrease cyclosporine concentrations should be avoided. Manufacturer lists antibiotics (e.g., nafcillin [Nallpen], rifampin [Rifadin]), anticonvulsants (e.g., carbamazepine [Tegretol], oxcarbazepine [Trileptal], phenobarbital [Luminal], phenytoin [Dilantin]), bosentan (Tracleer), octreotide (Sandostatin), orlistat (Alli, Xenical), terbinafine (Lamisil), ticlopidine (Ticlid), St. John’s wort, and sulfinpyrazone (Anturane). Other sources list sulfamethoxazole/trimethoprim; monitor levels and adjust cyclosporine dose as indicated to avoid transplant rejection. ■ Rifabutin (Mycobutin) may increase metabolism of cyclosporine; use care with concomitant use. ■ Cyclosporine inhibits CYP3A4 and the multidrug efflux transporter P-glycoprotein and may increase plasma concentrations of co-medications that are substrates of CYP3A4, P-glycoprotein, or organic anion transporter proteins. Cyclosporine reduces clearance and may increase blood levels of ambrisentan (Letairis), bosentan (Tracleer), dabigatran (Pradaxa), digoxin (Lanoxin), etoposide (VePesid), methotrexate, NSAIDs, prednisolone, repaglinide (Prandin), sirolimus (Rapamune), and other drugs. May decrease the volume distribution of digoxin and cause toxicity rather quickly. With concurrent use, monitor digoxin levels, reduce digoxin dose, or discontinue as indicated. ■ Concomitant use with NSAIDs, particularly in dehydrated patients, may potentiate renal dysfunction. ■ Avoid concurrent use with bosentan. ■ When coadministering ambrisentan with cyclosporine, the ambrisentan dose should not be titrated to the recommended maximum daily dose. ■ Cyclosporine may decrease the clearance of HMG-CoA reductase inhibitors (statins) such as atorvastatin (Lipitor), lovastatin (Mevacor), pravastatin (Pravachol), simvastatin (Zocor) and, rarely, fluvastatin (Lescol). Cases of myotoxicity (including muscle pain and weakness, myositis, and rhabdomyolysis) have been reported with concomitant use. Dose reduction of statins is indicated. Statins may be temporarily withheld or discontinued in patients with S/S of myopathy or potential for renal injury, including renal failure, secondary to rhabdomyolysis. ■ Coadministration with aliskiren (Tekturna) is not recommended. ■ May decrease mycophenolate (CellCept) levels. Monitor levels closely when cyclosporine is added or removed from a drug regimen containing mycophenolate. ■ Concurrent use of cyclosporine with imipenem-cilastatin (Primaxin) may increase CNS toxicity of both agents. ■ Potentiates nondepolarizing muscle relaxants (e.g., atracurium [Tracrium]); will prolong neuromuscular blockade. ■ Do not use potassium-sparing diuretics (e.g., spironolactone [Aldactone]); may increase risk of hyperkalemia. Use caution when coadministered with other potassium-sparing drugs (e.g., angiotensin-converting inhibitors [e.g., enalapril (Vasotec), lisinopril (Zestril)], angiotensin II receptor antagonists [e.g., losartan (Cozaar), valsartan (Diovan)]), potassium-containing drugs, and/or in patients on a potassium-rich diet. Hyperkalemia can occur. ■ May cause convulsions with high doses of methylprednisolone. ■ May be given in combination with steroids but has additive effects with other immunosuppressive agents; may increase risk of lymphoma. ■ Concurrent administration with sirolimus (Rapamune) increases blood levels of sirolimus. To minimize the effect on blood levels, administer sirolimus 4 hours after cyclosporine dose. ■ Elevations in SCr have been reported with coadministration of sirolimus and cyclosporine. Effect is usually reversible with cyclosporine dose reduction. ■ Serum levels may increase with chloroquine (Aralen). ■ Avoid use in psoriasis patients receiving other immunosuppressive agents or radiation therapy, including PUVA and UVB. Immunosuppression may be excessive. ■ May increase the plasma concentrations of repaglinide (Prandin), which increases the risk for hypoglycemia. Monitor blood glucose levels closely. ■ Concurrent use with nifedipine (Procardia) has caused gingival hyperplasia; avoid use in patients who develop gingival hyperplasia. ■ High doses of cyclosporine (e.g., IV doses of 16 mg/kg/day) may increase the exposure to anthracycline antibiotics (e.g., daunorubicin, doxorubicin, mitoxantrone) in cancer patients. ■ Monitor serum creatinine when used with NSAIDs in rheumatoid arthritis patients. ■ Vaccinations may be less effective. Avoid use of live virus vaccines in patients receiving cyclosporine. ■ Grapefruit juice may affect certain enzymes of the P450 enzyme system and should be avoided.
Side effects
The most common side effects include gum hyperplasia, hirsutism, hypertension, renal dysfunction, and tremor. Other side effects include acne, convulsions, cramps, diarrhea, encephalopathy, glomerular capillary thrombosis, headache, hepatotoxicity, hyperkalemia, hyperuricemia, hypomagnesemia, infection, leukopenia, lymphoma, microangiopathic hemolytic anemia, nausea and vomiting, paresthesia, skin rash, and thrombocytopenia. Hypersensitivity reactions including anaphylaxis have occurred. Stevens-Johnson syndrome and toxic epidermal necrolysis have occurred rarely.
Post-marketing:
Headache, including migraine; hepatotoxicity and liver injury, including cholestasis, hepatitis, jaundice, and liver failure with serious and/or fatal outcomes; isolated cases of pain in the lower extremities; JC virus–associated PML, sometimes fatal; and PVAN, especially due to BK virus infection, resulting in graft loss.
Antidote
Notify physician of all side effects. Most can be treated symptomatically. Drug may be decreased or discontinued or other immunosuppressive agents utilized. Consider reducing total immunosuppression in transplant patients who develop PML or PVAN; may place graft at risk. Discontinue infusion at the first sign of a severe hypersensitivity reaction. Treat hypersensitivity as indicated; may require oxygen, epinephrine (Adrenalin), antihistamines (e.g., diphenhydramine [Benadryl]), vasopressors (e.g., dopamine), corticosteroids, albuterol, IV fluids, and/or ventilation equipment. Nephrotoxicity, hepatotoxicity, encephalopathy (including PRES), or hematopoietic depression may require temporary reduction of dosage or permanent withholding of treatment. Dialysis is not effective in overdose.
Cytarabine 
(sye-TAIR-ah-bean)
ARA-C, Cytosar  , Cytosar-U
, Cytosar-U 
Antineoplastic (antimetabolite)
pH 5
Usual dose
Acute nonlymphocytic leukemia in adult and pediatric patients:
Single agent: 200 mg/M2/24 hr as a continuous infusion for 5 days. Repeat every 2 weeks. Combination chemotherapy: Dose is variable depending on specific regimen or protocol. Examples are 100 mg/M2/24 hr as a continuous infusion or 100 mg/M2 as an IV injection every 12 hours. Repeat daily on days 1 through 7. Another regimen uses 100 to 200 mg/M2 or 2 to 6 mg/kg/24 hr as a continuous infusion or equally divided into 2 or 3 doses and given by IV injection or intermittent infusion. Given for 5 to 10 days. Maintain treatment until therapeutic effect or toxicity occurs. Modify on a day-to-day basis for maximum individualized effectiveness.
Acute myelocytic leukemia or erythroleukemia in adult and pediatric patients:
As a single agent, 100 to 200 mg/M2/24 hr or 3 mg/kg/24 hr for 5 to 10 days as a continuous infusion or in divided doses by IV injection. Total dose is 1,000 mg/M2. Repeat every 2 weeks.
Dose adjustments
Dose (mg/kg) based on average weight in presence of edema or ascites. ■ Dose reduction may be indicated in impaired hepatic or renal function. ■ See Precautions/Monitor. ■ Usually used with other antineoplastic drugs in specific doses to achieve tumor remission. ■ Withhold or modify dose based on degree of bone marrow suppression; see Monitor.
Dilution
Specific techniques required; see precautions.
Some preparations are liquid and do not require reconstitution or each 100 mg must be reconstituted with 5 mL (500 mg with 10 mL) of SWFI with benzyl alcohol 0.9%. Solution pH about 5. May be given by IV injection as is or further diluted in NS or D5W and given as an infusion. IV injection should be through a free-flowing IV tubing. Use only clear solutions.
Storage:
Stable at room temperature for 48 hours.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Daunorubicin (Cerubidine), gentamicin, hydrocortisone sodium succinate (Solu-Cortef), lincomycin (Lincocin), methotrexate, methylprednisolone (Solu-Medrol), mitoxantrone (Novantrone), ondansetron (Zofran), potassium chloride (KCl), sodium bicarbonate, vincristine.
Y-site:
Amifostine (Ethyol), amphotericin B cholesteryl (Amphotec), anidulafungin (Eraxis), aztreonam (Azactam), cladribine (Leustatin), doxorubicin liposomal (Doxil), etoposide phosphate (Etopophos), filgrastim (Neupogen), fludarabine (Fludara), gemcitabine (Gemzar), gentamicin, granisetron (Kytril), hydrocortisone sodium succinate (Solu-Cortef), idarubicin (Idamycin), linezolid (Zyvox), melphalan (Alkeran), methotrexate, methylprednisolone (Solu-Medrol), ondansetron (Zofran), paclitaxel (Taxol), pemetrexed (Alimta), piperacillin/tazobactam (Zosyn), propofol (Diprivan), sargramostim (Leukine), sodium bicarbonate, teniposide (Vumon), thiotepa, vinorelbine (Navelbine).
Rate of administration
IV injection:
Each 100 mg or fraction thereof over 1 to 3 minutes.
IV infusion:
Single daily dose properly diluted over 30 minutes to 24 hours, depending on amount of infusion solution and dosage regimen.
Actions
An antimetabolite and pyrimidine antagonist that interferes with the synthesis of DNA. Cell cycle specific for S phase. Through various chemical processes this deprivation acts more quickly on rapidly growing cells and causes their death. Cytotoxic and cytostatic. A potent bone marrow suppressant. Crosses the blood-brain barrier. Serum half-life averages 1 to 3 hours. Metabolized in the liver and excreted in the urine.
Indications and uses
Induction and maintenance of remission in acute nonlymphocytic leukemia in adults and pediatric patients. Also used for treatment of acute lymphocytic leukemia (ALL), the blast phase of chronic myelocytic leukemia, and acute myelocytic leukemia (AML). ■ Is used intrathecally in the treatment of meningeal leukemia. A liposomal formulation (DepoCyt) is available for intrathecal use only, lipofoam molecules contained in this product are much too large for IV use.
Contraindications
Hypersensitivity to cytarabine, pre-existing drug-induced bone marrow suppression.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of a physician specialist in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Remissions induced by cytarabine are brief unless followed by maintenance therapy. ■ Use caution with impaired liver or renal function. ■ Severe GI, pulmonary, or CNS toxicity has occurred with experimental cytarabine regimens. Toxicities are different (e.g., reversible corneal toxicity, hemorrhagic conjunctivitis, cerebral and cerebellar dysfunction, severe GI ulceration) from those seen with conventional therapy. Deaths have been reported. ■ Benzyl alcohol may cause a fatal “gasping syndrome” in premature infants.
Monitor:
Leukocyte and platelet counts should be monitored daily. ■ During induction therapy, WBC depression is biphasic with the first nadir occurring at days 7 to 9 and a deeper fall at days 15 to 24. Platelet depression begins around day 5 and reaches a nadir at days 12 to 15. ■ Hold or modify therapy for platelet count less than 50,000 or polymorphonuclear granulocytes less than 1,000 cells/mm3. Restart therapy when bone marrow recovery is confirmed. ■ Monitor bone marrow, liver, and renal function at regular intervals during therapy. ■ Higher doses tolerated by IV injection compared with IV infusion, but the incidence and intensity of nausea and vomiting are increased. ■ Prophylactic administration of antiemetics recommended. ■ Be alert for signs of bone marrow suppression, bleeding, infection, or neurotoxicity. These side effects are dose- and schedule-dependent. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. ■ Monitor uric acid levels; maintain hydration; allopurinol may be indicated.
Patient education:
Nonhormonal birth control recommended. ■ See Appendix D, p. 1333. ■ Promptly report early signs of neurotoxicity (e.g., ataxia, confusion, lethargy).
Maternal/child:
Category D: avoid pregnancy. May produce teratogenic effects on the fetus, especially during the first trimester. ■ Discontinue breast-feeding. ■ See Drug/Lab Interactions.
Elderly:
Consider age-related organ impairment; toxicity may be increased.
Drug/lab interactions
May inhibit digoxin absorption. ■ Do not administer any live virus vaccines to patients receiving antineoplastic drugs. ■ May cause acute pancreatitis in patients who previously received l-asparaginase. ■ May antagonize action of gentamicin against Klebsiella. ■ May antagonize antifungal actions of flucytosine (Ancobon). ■ Clearance decreased and toxicity increased with nephrotoxic agents (e.g., aminoglycosides [gentamicin]); may cause neurotoxic symptoms (e.g., ataxia, confusion, lethargy). ■ Concurrent use of high doses of cytarabine with cisplatin may increase ototoxicity.
Side effects
Abdominal pain, bone marrow suppression (e.g., anemia, leukopenia, thrombocytopenia), bone pain, cardiomyopathy, chest pain, conjunctivitis, diarrhea, esophagitis, fever, hepatic dysfunction, hypersensitivity reactions, hyperuricemia, malaise, megaloblastosis, mucosal bleeding, myalgia, nausea, oral ulceration, pancreatitis, peripheral motor and sensory neuropathies, rash, stomatitis, thrombophlebitis, vomiting. Higher than usual dose regimens may cause severe coma, GI ulcerations and peritonitis, personality changes, pulmonary toxicity, somnolence, or death.
Antidote
Notify the physician of all side effects. Most will be treated symptomatically. Some toxicity is necessary to produce remission. Discontinue the drug for serious bone marrow suppression. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. Drug must be restarted as soon as signs of bone marrow recovery occur, or its effectiveness will be lost. Use corticosteroids for cytarabine syndrome (fever, myalgia, bone pain, occasional chest pain, maculopapular rash, conjunctivitis, malaise). Usually occurs in 6 to 12 hours after administration. Continue cytarabine if patient responds to corticosteriods. There is no specific antidote; supportive therapy as indicated will help to sustain the patient in toxicity.
Cytomegalovirus immune globulin intravenous (human)  *
*
(sigh-toh-meg-ah-lo-VIGH-rus ih-MUNE GLAW-byoo-lin)
CMV-IGIV, CytoGam
Passive immunizing agent
Antibacterial
Antiviral
Usual dose
150 mg/kg is the maximum recommended dose per infusion.
Kidney transplant:
150 mg/kg of body weight as an IV infusion. This initial dose must be given within 72 hours of transplant. Additional infusions of 100 mg/kg are given at 2, 4, 6, and 8 weeks posttransplant, then reduced to 50 mg/kg at 12 and 16 weeks posttransplant.
Heart, liver, lung, and pancreas transplants:
150 mg/kg of body weight as an IV infusion. This initial dose must be given within 72 hours of transplant. Consider use in combination with ganciclovir (Cytovene) 10 mg/kg/day for 14 days. Additional infusions of cytomegalovirus IGIV containing 150 mg/kg are given at 2, 4, 6, and 8 weeks posttransplant, then reduced to 100 mg/kg at 12 and 16 weeks posttransplant.
Dilution
Absolute sterile technique required; contains no preservatives. Available in 20- and 50-mL vials (50 mg/mL); multiple vials may be required. Use only if clear and colorless. Enter vial only once and initiate infusion within 6 hours. Must be completely infused within 12 hours of dilution. See Rate of Administration.
Filters:
Use of an in-line filter (pore size 15 microns [0.2 microns acceptable]) is required; see Rate of Administration.
Storage:
Store dry powder in refrigerator between 2° and 8° C (35° to 46° F).
Compatibility
Administration through a separate infusion line recommended. If absolutely necessary, may be piggybacked in a pre-existing line containing NS, 1/2NS, dextrose 2.5%, 5%, 10%, or 20% in water or saline. Do not dilute CMV-IGIV more than one part to two parts of any of these solutions.
Rate of administration
Use of an in-line filter (pore size 15 microns [0.2 microns acceptable]) and a constant infusion pump (e.g., IVAC) is required. Begin with a rate of 15 mg/kg/hr. May be increased to 30 mg/kg/hr in 30 minutes if no discomfort or adverse effects. May be increased in another 30 minutes to 60 mg/kg/hr if no discomfort or adverse effects. Do not exceed the 60 mg/kg/hr rate or allow the volume infused to exceed 75 mL/hr regardless of mg/kg/hr dose. Slow rate of infusion at onset of patient discomfort or any adverse reactions. Infusion must be complete within 12 hours of dilution. Subsequent doses may be increased at 15-minute intervals using the same mg/kg/hr rates and adhering to the volume maximum of 75 mL/hr.
Actions
A sterile solution of immunoglobulin G (IgG). Derived from pooled adult human plasma selected for high titers of antibody for cytomegalovirus (CMV). Purified by a specific process. Can raise the relevant antibody levels sufficiently to attenuate or reduce the incidence of serious CMV disease. Antibody levels will last 2 to 3 weeks. Recent studies of combined prophylaxis with CMV-IGIV and ganciclovir have shown reductions in the incidence of serious CMV-associated disease in CMV-seronegative recipients of CMV-seropositive organs below that expected from one drug alone.
Indications and uses
Prophylaxis of CMV disease associated with transplantation of kidney, heart, liver, lung, and pancreas. In transplants of these organs other than kidney from CMV-seropositive donors to seronegative recipients, prophylactic CMV-IGIV should be considered in combination with ganciclovir.
Contraindications
History of a prior severe reaction associated with any human immunoglobulin preparations. Individuals with selective immunoglobulin A deficiency may develop antibodies to IgA and are at risk for anaphylaxis.
Precautions
75% of untreated recipients would be expected to develop CMV disease. Use of CMV-IGIV has effected a 50% reduction in this disease rate. Effective results have been obtained with a variety of immunosuppressive regimens (e.g., combinations of azathioprine, cyclosporine, prednisone). ■ A fatal CMV infection occurred even with ganciclovir treatment in one patient, who inadvertently missed a single injection.
Monitor:
Continuous monitoring of vital signs is preferred. Must be monitored before infusion, at every rate change, at the midpoint, at the conclusion, and several times after completion. ■ All supplies for emergency treatment of acute anaphylatic reaction must be available; see Antidote.
Patient education:
Adherence to the prescribed regimen is imperative.
Maternal/child:
Category C: safety for use during pregnancy or breast-feeding not established. Use only if clearly needed.
Drug/lab interactions
Defer vaccination with any live virus vaccine (e.g., measles, mumps, rubella) until 3 months after CMV-IGIV administration.
Side effects
Incidence related to rate of administration; back pain, chills, fever, flushing, hypotension, muscle cramps, nausea, vomiting, wheezing. Hypersensitivity reactions, including anaphylaxis, are possible.
Antidote
With onset of any minor side effect, reduce rate of infusion immediately or discontinue temporarily. Discontinue CMV-IGIV if symptoms persist, and notify the physician. May be treated symptomatically and infusion resumed at a slower rate if symptoms subside. Discontinue CMV-IGIV if hypotension or anaphylaxis occur and treat immediately. Epinephrine (Adrenalin), diphenhydramine (Benadryl), oxygen, vasopressors (e.g., dopamine), corticosteroids, and ventilation equipment must always be available. Resuscitate as necessary.

Full access? Get Clinical Tree


