Chapter 34. Caring for children with diabetes and other endocrine disorders
Philomena Morrow, Sue Courtman and Liz Gormley-Fleming
LEARNING OUTCOMES
• Understand the role and function of the endocrine system.
• Appreciate how disordered function affects the child.
• Understand the care requirements of the child and family in relation to specific endocrine disorders.
• Understand the specialist nature of caring for a child with diabetes.
• Recognise the importance of health promotion/education in the care of the child and family with an endocrine disorder.
• Appreciate the need for continuing care and support required by the child and family with an endocrine disorder.
Endocrine system
The endocrine system is a chemical communication system that provides the means to control a number of physiological processes within the body. As it is a communication network, the endocrine system contains transmitters, signals and receivers that are called hormone-producing cells, hormones and receptors. The endocrine system consists of a number of distinct glands and some tissues in other organs. The endocrine glands (Fig. 34.1) are:
• pituitary gland
• thyroid gland
• four parathyroid glands
• two adrenal glands
• pancreatic islets (also known as the islets of Langerhans)
• pineal gland
• thymus gland
• two ovaries in the female
• two testes in the male.
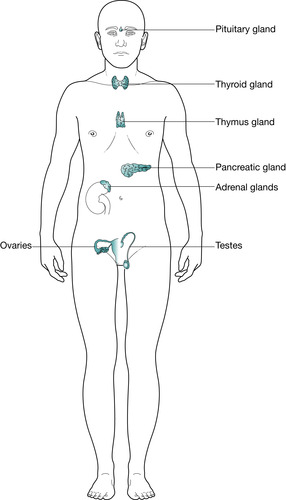 |
| Fig. 34.1 The glands within the body. |
Endocrine glands secrete hormones directly into the bloodstream, whereas exocrine glands pass their secretions directly into body cavities. The hormones released by endocrine glands are carried in the bloodstream to their target organ, where they are active. The activity of a number of endocrine glands is regulated through the activity of the hypothalamus.
The hypothalamus
Nine different substances have been identified as hypothalamic hormones that either stimulate or inhibit the release of anterior and posterior hormones (Table 34.1). These hormones are formed in the median eminence of the hypothalamus (the area that is connected to the pituitary gland by the pituitary stalk).
 PowerPoint
PowerPoint
 PowerPoint
PowerPoint
PowerPointAccess the companion PowerPoint presentation and revise your knowledge in relation to hormone structure, action of hormones, regulation and synthesis and metabolism and excretion of hormones.
PowerPointAccess the companion PowerPoint presentation and look at the discussion on hypopituitarism and hyperpituitarism including pituitary tumours to enhance your knowledge.
You are also advised to revise the section on growth hormone production, regulation and synthesis before proceeding to the next section of this chapter on growth hormone deficiencies.
| Hypothalamus | ||
| Synthesis of posterior pituitary hormones and transport via nerve axons | Hypothalamic hormones carried in hypothalamohypophyseal portal system | |
| Pituitary gland | ||
| Posterior pituitary (neurohypophysis) | Middle lobe | Anterior lobe (adenohypophysis) |
| Oxytocin | Melanocyte-stimulating hormones | Trophic hormones:ACTH, adrenal cortex hormone |
| Vasopressin (antidiuretic hormone) | ||
| Thyroid stimulating hormone (TSH) | ||
| Growth hormone (GH) | ||
| Follicle stimulating hormone (FSH) | ||
| Luteinising hormone (LH) | ||
| Prolactin (PRL) | ||
Pituitary gland
The gland develops from the merging of different tissues (Fig. 34.2).
 |
| Fig. 34.2 The pituitary gland. |
Anterior pituitary gland
Originates from an upgrowth of glandular epithelium from the pharynx and is known as the adenohypophysis. This is only linked to the brain via the venous hypothalamohypophyseal portal system. This network transports blood from the hypothalamus to the anterior pituitary, thereby transporting releasing and inhibiting hormones secreted by the hypothalamus. These hormones influence secretion and release of other hormones formed in the anterior pituitary (Fig. 34.3):
• Corticotrophin-releasing hormone: controls release of tropic hormones, such as adrenocorticotrophic hormone (ACTH)
• Thyrotrophin-releasing hormone (also known as thyroid-stimulating hormone)
• Growth-hormone-releasing factor
• Gonadotrophin-releasing hormone
• Somatostatin-releasing hormone (also known as growth-hormone-release inhibiting factor)
• Prolactin-inhibiting factor
• Melanocyte-stimulating hormone (MSH).
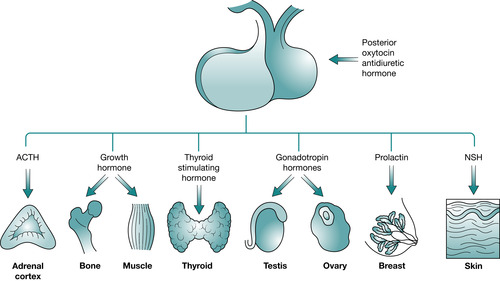 |
| Fig. 34.3 Anterior pituitary hormones. |
Posterior pituitary gland
This is derived from a downgrowth of nervous tissue from the base of the brain and is known as the neurohypophysis. It has nervous connections with the hypothalamus, down which the antidiuretic hormone (ADH) and oxytocin pass. ADH and oxytocin are synthesised in the nerve cell bodies of the hypothalamus, transported along the axons, and then stored and secreted by vesicles within the axon terminals. Their release is triggered by nerve impulses from the hypothalamus.
Disorders affecting the pituitary gland
Disorders affecting the pituitary gland result in disruption in regulation of hormone secretion. Symptoms vary depending on the location of the disorder, and are related to a disturbance in production of a specific hormone or group of hormones and relate to the role they play in maintaining health and development.
Hypopituitarism
This term denotes subnormal pituitary hormone production (Parks 2002). It is generally a result of disorders of the anterior pituitary and may involve one or more of the hormones produced (Table 34.2). Deficiencies of anterior hormone production are due to primary disease or disorder of the anterior pituitary. Secondary hypopituitarism, which is attributed to dysfunction of the hypothalamus, affects the synthesis and release of releasing hormones and release inhibiting hormones.
 PowerPoint
PowerPoint
PowerPointAccess the companion PowerPoint presentation and refer to the discussion on growth hormone for detailed information on assessment and diagnosis in relation to family history, child’s history, radiographic and imaging findings, and laboratory findings.
| Hypopituitarism | Hypopituitarism |
|---|---|
| Growth hormone: somatic growth retardation | Somatic growth acceleration Adrenal hyperfunction |
| Adrenocorticotrophic hormone (ACTH): adrenal hypofunction | Hyperthyroidism Precocious puberty/retarded sexual development |
| Thyroid hormone: hypothyroidism | |
| Follicle stimulating and luteinising hormone: absence/regression of secondary sexual characteristics | Prolactin: stops menstruation |
| Prolactin |
Factors contributing to pituitary hyposecretion include congenital hypopituitarism due to genetic abnormalities or other developmental defects. Congenital hypopituitarism is mainly related to growth hormone deficiency. Disordered pituitary hormone secretion that develops after birth is referred to as acquired hypopituitarism (Parks 2002). This may have resulted from central nervous system trauma, meningitis or encephalitis, vascular abnormalities or haemorrhage, brain tumours, pituitary tumours and/or radiation therapy.
Hyperpituitarism
Hyperpituitarism is a disorder in which excessive secretion of growth hormone increases the growth rate. Pituitary tumours that secrete hormones produce characteristic symptoms of excessive hormone levels. This is rare in children. Symptoms may range from complaints such as listlessness or restlessness to more severe symptoms such as headaches, vomiting or dizziness. In addition, problems due to an increase in any of the hormones produced may give rise to growth, adrenal or sexual dysfunction (see Table 34.2).
Hormonal abnormalities occurring as a result of pituitary tumours or defects will be discussed under specific headings. Oversecretion of growth hormone is usually caused by a pituitary adenoma.
Precocious puberty
Precocious puberty is defined as the onset of secondary sex characteristics by the age of 8.5 years in females and 9.5 years in males (Rudolf & Levene 2006). It is either gonadotrophin dependant or gonadotrophin independent. It is more likely to occur in females and may be idiopathic in origin and is likely to be due to the premature onset of normal puberty. It is recognised today that pubertal development has started to occur earlier due to improvements in nutrition and socioeconomic conditions. While it is rare it males, it is far more likely to be due to organic causes. The causes of precocious puberty may be:
• idiopathic/familial tendency
• CNS abnormalities
• acquired – following surgery, trauma, irradiation
• congenital abnormalities – hydrocephalus
• tumours
• adrenal – congenital adrenal hyperplasia, tumours
• ovarian tumours
• testicular tumours
• exogenous sex steroids
• hypothyroidism.
Diagnosis
Diagnosis is based on physical examination, blood analysis, bone age measurements, ultrasonography and radiological imaging – MRI or CT scanning.
Treatment
This is dependent on the cause and may necessitate surgery, chemotherapy or irritation if a tumour is diagnosed. For the female with idiopathic precocious puberty treatment will depend on the age of diagnosis. Monitoring of growth patterns is essential. Treatment is usually administration of gonadotrophin releasing hormone subcutaneously, intramuscularly or intranasally for those cases that are gonadotrophin dependent.
Role of the nurse
The aim of nursing care should focus on the education of the child and parent about the condition, medication administration and to provide emotional support for the family. There may be psychological and behavioural difficulties associated with the early progression into puberty. It must be emphasised to the family that the child is achieving their other developmental milestones – cognition, emotional development and social development – according to their chronological age even if their physical development and appearance is advanced.
Growth hormone deficiencies
Growth hormone deficiency exists when growth hormone is absent or is produced in inadequate amounts to support normal growth. If growth hormone deficiency occurs in combination with one or more other pituitary hormone deficiencies the condition is related to hypopituitarism, as described earlier.
Growth hormone deficiency can be congenital, resulting from deficiency of hypothalamic growth hormone releasing hormone that may be associated with defects such as septo-optic dysplasia or primary pituitary disorders such as defects in the growth hormone gene. Most congenital cases of growth hormone deficiency are currently considered idiopathic (Cuttler 2002), which is the most common form, accounting for approximately 50–70% of cases (NICE 2002). Fetal growth is growth hormone independent and therefore infants with congenital growth hormone deficiency are born normal size and weight. However, growth hormone deficiency may present in the newborn period with hypoglycaemia, and prolonged jaundice. These symptoms are often significant early diagnostic features. Growth hormone deficiency becomes evident only during the first years of life, when linear growth begins to slow and is the 3rd percentile or less by the age of 1 year.
Acquired growth hormone deficiency may be the result of injury, infection, inflammatory and granulomatous disease, radiation or tumours of the pituitary gland and/or the hypothalamus; it may become evident during infancy or childhood. Failure to grow normally may also be a key feature of other underlying medical conditions, e.g. chronic renal insufficiency, Turner syndrome, Prader–Willi syndrome, Down syndrome, neurofibromatosis and as a result of chronic disease. Among children who are of very short stature (i.e. at least 3 standard deviations below the population mean), 25% have growth hormone deficiency (NICE 2002).
Principles of nursing care and management
Care and management will be based on the assessment of individual needs of the child and in collaboration with the child and family (Table 34.3). The deficiency is identified by serum analysis of growth hormones. Brain imaging is also required to identify underlying pathology. A detailed history of the child’s physical status and social situation needs to be ascertained.
| Characteristics of child with growth hormone deficiency | Nursing considerations |
|---|---|
| Short stature | Accurate assessment and monitoring of height and weight |
| Delayed growth of all body parts | Accurate recording/plotting height and weight on appropriate growth chart |
| Delayed skeletal maturation | Early recognition of deviation in height and weight pattern |
| Immature facial appearance | Accurate detailed assessment of child’s health and health problems |
| Increased subcutaneous fat | Assessment of family history |
| In prolonged growth failure the child will be shorter than children of the same age | Referral for medical consultation and assisting and supporting the child/family during medical examination, radiological surveys and endocrine studies |
| Hypoglycaemia may be present particularly in young children |
In children with growth hormone insufficiency, exogenous (biosynthetic) growth hormone (somatropin) is administered by subcutaneous injection; the dose is increased during the period of adolescence. It is continued until the child attains their final height (Rudolf & Levene 2006). The recommended dose varies according to the child’s condition and is self-administered at home 6–7 times a week. Biosynthetic human growth hormone has been licensed for use in the UK for long-term treatment of children who have growth failure due to inadequate secretion of normal endogenous growth hormone (NICE 2002). A variety of growth hormone preparations and devices are available, knowledge of which is important if the nurse is to help the child and family choose the most suitable product and device. Although treatment enables many children to reach their adult height potential, this may mean many years of treatment for the child.
Establishing and maintaining optimum therapy both at the outset and over what may be a long period of time may be extremely challenging for many families (Nairn & Moore 2002). The initial and ongoing education of the child and family and the monitoring and maintenance of long-term compliance to achieve the best possible outcomes are important aspects within the role of the children’s nurse. In the presence of secondary growth hormone deficiencies the underlying lesion needs to be treated and prognosis is related to the underlying lesion.
Side effects of growth hormone therapy are rare but can include headache, visual problems, nausea and vomiting, fluid retention (peripheral oedema), arthralgia, myalgia, paraesthesia, antibody formation, hypothyroidism and reactions at injection site. Particular attention should be paid to treating children with risk factors associated with diabetes mellitus and slipped capital epiphyses.
Storage and preparation of medication, as well as choosing and alternating the injection site are important aspects of the education process. Timing of administration of growth hormone may be dependent on family routines. The evening is usually the recommended time as this mimics the pattern of normal production of growth hormone. (Hockenberry et al 2003). Compliance to treatment may be enhanced by enabling the child to take control over administration of growth hormone, which may be achieved by the use of the pen injector. The best results are seen in those that are treated early before psychological effects occur.
 Evidence-based practice
Evidence-based practice
 Reflect on your practice
Reflect on your practice
 Activity
Activity
 Activity
Activity
Evidence-based practiceGrowth hormone therapy can increase short-term growth and improve (near) final height (Bryant et al 2004).
Reflect on your practice• Explain to a parent the technique required to administer a subcutaneous injection.
• Review the advice regarding proper injecting technique for growth hormone at: http://www.heightmatters.org.uk
ActivityChildren and parents need to understand the importance of complying with treatment regimes and to be competent in administration. This requires that the child and family are educated regarding the rationale for treatment and adverse effects.
ActivityReview a growth chart for a boy and a girl from birth to 18 years. Identify the nurse’s responsibilities in relation to the assessment of the following:
• Weight
• Standing height
• Supine length
• Head circumference.
Review the PowerPoint presentation for measurement technique for standing height (Hall 2000).
Child and family resources
Raising awareness of the guidance on the use of human growth hormone in children with growth failure (NICE 2002; website: http://www.nhsdirect.nhs.uk) and encouraging parents to discuss this guidance with their doctor will provide further encouragement to comply with treatment. The Human Growth Foundation is a national organisation of parents who provide education and guidance on the physical, psychological and social development of children with growth problems. This resource may be beneficial in providing parents and children with additional support and advice. Good nutrition and adequate rest is vital when promoting growth. A well-balanced diet with appropriate calorie and protein intake needs to be encouraged.
Children’s nurses have a significant role in the routine monitoring of growth and the assessment of growth disorders. The development of good history-taking skills and an accurate, repeatable measurement are central to the success of growth assessment and evaluation. Measuring height is subject to error as a result of poor technique, variations between instruments and observers and diurnal variation (Hall 2000). The child should be measured by the same observer using the same measuring instrument if possible to maintain accuracy and consistency. Parental height should also be measured with the mid parental centile and target height plotted on the percentile chart (Drake & Kelnar 2006).
During therapy, the child’s growth needs to be monitored against expected growth on standard growth charts to assess ongoing response to treatment (Table 34.4). Explain to the parents that developmental expectations are the same for a child with growth hormone deficiencies as they are for a child who is developing normally. Expected growth may be based on parental height. It is recommended that treatment should be discontinued if the child’s growth velocity is less than 50% from baseline in the first year of therapy, i.e. if extra height gain is not at least half the height gain in the year before treatment began. Persistent problems with adherence to treatment should also be taken into account as part of the re-evaluation process (NICE 2002).
 Evidence-based practice
Evidence-based practice
 Activity
Activity
Evidence-based practiceHeight is greatest on getting up in the morning – up to 2 cm can be lost over the whole day. Measurements made at different times of the day can significantly affect the measured height and, thus, the estimated rate of growth (Hall 2000).
ActivityReview Chapter 46 on caring for a child requiring palliative care:
• Discuss how this information may be relevant to the care of the child and family in the above situation in relation to bereavement and loss.
| Age | Growth in height |
|---|---|
| 0–6 months | 18–26 cm per year |
| 6–12 months | 15–28 cm per year |
| 1–2 years | 10–13 cm per year |
| 2–3 years | 7–10 cm per year |
| 3–4 years | 5–8 cm per year |
| 4–5 years | 5 cm per year |
Recognition of the complexity of problems experienced by the child and family in relation to growth deficiency and/or short stature is necessary if physical and psychosocial dilemmas are to be overcome. Children who are small for their age may have problems with friends, teachers and parents, who tend to treat them as though they were younger and have reduced expectations of them. In turn, children may not act their age because it is not expected from them and thus experience problems with self-esteem and ability to interact appropriately with others. The child or young person should be encouraged to dress age appropriate rather than size and treated in an age-appropriate manner. Those involved in the care of these children should be aware of the importance of emphasising abilities and strengths rather than physical size.
 PowerPoint
PowerPoint
PowerPointAccess the companion PowerPoint presentation and revise your knowledge on the production and regulation of secretion of the thyroid hormones before proceeding to the next section of this chapter on disorders of thyroid function.
Disorders of thyroid function
The function of the thyroid gland is to regulate the cellular metabolism. Thyroxine T4, tri-iodothyronine T3 and calcitonin are hormones secreted by the thyroid. These hormones are responsible for normal development of the muscular, skeletal and nervous systems. Disturbance in the secretory pathway of the thyroid hormones may result in an increase or decrease in production. Some of the most common endocrine disorders are disorders of the thyroid gland. The two main disorders are hypothyroidism and hyperthyroidism.
Hypothyroidism
Hypothyroidism is either congenital or acquired (juvenile hypothyroidism). It is due to a deficiency in the secretion of thyroid hormones. Congenital hypothyroidism is relatively common and occurs in approximately 1 in 4000 births (Lissauer & Clayden 2007). Early detection is imperative as this is one of the few preventable causes of severe learning difficulties. This is achieved by neonatal screening. Treatment is lifelong with oral replacement of thyroxine. Cause of congenital hypothyroidism are:
• hereditary defects in thyroid hormone synthesis that may be associated with maternal administration of antithyroid drugs during pregnancy (McCance & Huether 2006)
• iodine deficiency – common worldwide but rare in the UK; preventable by iodination of salt in the maternal diet
• thyroid stimulating hormone (TSH) deficiency.
Acquired juvenile hypothyroidism
Juvenile hypothyroidism may be primary or secondary. This is more common in females (Ball & Bindler 2006). Primary causes include:
• defective hormone synthesis resulting from acute thyroiditis caused by bacterial infection
• subacute non-bacterial inflammation associated with viral infections
• autoimmune (circulating antithyroid antibodies) thyroiditis occurring as a result of Hashimoto disease is the most common cause of juvenile hypothyroidism in children (Roth et al 1997)
• an endemic of iodine deficiency or antithyroid drugs, or loss of thyroid tissue can be contributory factors in hypothyroidism.
Secondary hypothyroidism is due to insufficient stimulation of the thyroid gland by thyroid stimulating hormone as a result of hypothalamic or pituitary disorders.
Other autoimmune disorders may develop, e.g. type 1 diabetes mellitus. This is more common in children with Down and Turner syndrome. Addison’s disease may also occur in some families (Lissauer & Clayden 2006).
 www
www
 www
www
wwwReview the UK process measures, standards and supporting implementation guidelines for newborn blood spot screening. These are available on the website of the UK Newborn Screening Programme Centre at:
wwwObtain further information on thyroxine and recommended dosages at:
Parent information is available at:
Information for children is available at:
Nursing assessment and diagnosis
Routine neonatal biochemical screening, blood spot test, must be performed on all infants either prior to discharge from hospital or in the community. This will identify most infants that are affected by this disorder.
In children under the age of 2 the symptoms may not be as clear as those associated with hypothyroidism therefore nursing assessment and ongoing monitoring of the neonate with presenting problems is vital to ensure early diagnosis of the condition. Serial measurement of weight, length and head circumference is an important aspect of assessment. Presenting symptoms depend on extent of dysfunction and age at onset. However, if undetected and left untreated in early childhood, permanent defects can arise, for example, mental retardation, deafness, deceleration in growth and other nervous system disorders. At birth there may be difficulty in identifying hypothyroidism. Hypothermia, delay in passing meconium and neonatal jaundice may be significant signs and require to be investigated immediately.
Clinical manifestations of hypothyroidism may not be evident until after 4 months of age. Signs and symptoms may include:
• failure to thrive and feeding problems
• a hoarse cry and protruding tongue caused by myxoedema of oral tissue and vocal cords
• the child may present with constipation and abdominal distension due to hypotonia of abdominal muscles
• umbilical hernia may also be present
• a subnormal temperature, excessive sleeping, slow pulse and a cold, mottled skin may be present
• puffy eyes and loss of the eyebrows may be present
Often the baby is seen as being a ‘good’ baby, as the baby is often quiet. Skeletal growth may be decelerated due to impaired synthesis of protein, poor absorption of nutrients and lack of bone mineralisation. Enlargement of the thyroid gland, known as goitre, may be evident, particularly in older children. Puberty may be delayed and obesity is often present. If the condition remains undiagnosed and untreated, the child will be dwarfed with short limbs (cretinism) as well as presenting with delayed intellectual development.
Care and management
Once diagnosis is confirmed, treatment is replacement therapy, which is prescribed according to the child’s hormonal levels. Treatment is required on a life-long basis. Regular estimates of hormone levels are essential to prevent development of hyperthyroidism. In addition, regular monitoring of growth is necessary throughout childhood. Assessment of cognitive development should also be undertaken. Parental education (and the child depending on their age) is essential as they need to demonstrate an understanding of the disorder and treatment required.
 Scenario
Scenario
 PowerPoint
PowerPoint
 Activity
Activity
ScenarioJune, a 12-year-old who presented with a history of weight loss, anxiety, disturbance in sleep and mood changes, was diagnosed as having Graves’ disease:
• When undertaking June’s nursing assessment, what specific information would you be required to obtain?
• What specific requirements would need to be considered when planning June’s care?
A discussion of this scenario appears at the end of the chapter.
PowerPointAccess the companion PowerPoint presentation and review regulation of adrenal and renal function before proceeding with the next section of this chapter on disorders of adrenal function.
ActivityReview Chapter 45 on caring for a critically ill child:
• Discuss how this information is relevant to the care of the child with acute adrenal insufficiency.
Hyperthyroidism
Hyperthyroidism is a condition in which the thyroid hormones exert greater than normal responses. Generally there is excess secretion of thyroid hormones, which might be associated with acute, subacute or chronic thyroiditis, tumours of the thyroid, or other tumours of the pituitary (which secretes thyroid-stimulating hormone). This leads to an increase in basal metabolic rate, cardiac function, gastrointestinal function, weight loss, and metabolism of fats, proteins and carbohydrates. While rare in childhood it is most common in teenage girls. Specific diseases that cause hyperthyroidism include Graves’ disease and toxic multinodular goitre.
Graves’ disease is the most common cause of hyperthyroidism in children. Although the exact cause is unknown, it is thought to be associated with autoimmune abnormalities that cause enlargement and thus hyperfunction of the thyroid gland.
Congenital hyperthyroidism may occur in infants of mothers who have Graves’ disease as a result of transplacental transfer of immunoglobulins (Ball & Bindler 2006)
Assessment and diagnosis
Hyperthyroidism is four times more common in girls than boys and most frequently presents in childhood between ages of 12 and 14 years. The condition usually presents with a history in deterioration in school performance.
The other clinical features of hypothyroidism will vary according to the amount and length of hypersecretion. The onset is subtle thus diagnosis may not be reached for a length of time. Weight loss, diarrhoea, rapid growth in height, tachycardia, tremors, increase in appetite, anxiety, learning difficulties/behaviour problems and goitre may be present. Eye signs are uncommon in children but exophthalmos may be evident (Lissaeur & Clayden 2007).
Diagnosis is achieved by laboratory analysis of thyroxine and tri-iodthyronine levels. A thyroid scan will also be performed. Antithyroid antibodies may also be identified in laboratory analysis.
The aim of treatment is to inhibit excessive secretion of the thyroid hormones. First-line treatment is oral medication and this is generally for a 2-year period. Adjunct therapy may also be required, e.g. beta-blockers. Radio-iodine treatment may follow oral medication. Surgery may also be considered if other treatment modalities are unsuccessful.
Nursing care
The aim of nursing care of the child with hyperthyroidism should focus on the education of the child and their parents. They need to be aware of the need to promote rest, the importance of compliance with treatment to avoid relapse and to provide emotional support. Pre- and postoperative care for the child undergoing subtotal or total thyroidectomy is very specific but the general principles of pre- and postoperative care still apply.
Disorders of adrenal function
Disorders of the adrenal cortex (Fig. 34.5) are related to either hyperfunction or hypofunction (Table 34.5).
 Evidence-based practice
Evidence-based practice
Evidence-based practicePuberty imposes greater difficulty in achieving and maintaining adrenal suppression despite optimal doses of substitution therapy (Charmandari et al 2002):
• Discuss.

 |
| Fig. 34.5 The adrenal gland. |
| Hyperfunction of adrenal cortex | Hypofunction |
|---|---|
| Cushing’s syndrome: increased levels of circulating cortisol | Acute adrenocortical insufficiency |
| Chronic adrenocortical insufficiency: Addison’s disease | |
Congenital adrenal hyperplasia: increased secretion of adrenal androgens and oestrogens leading to virilisation or feminisation Primary hyperaldosteronism Hyperfunction of adrenal medulla Phaeochromocytoma: increased secretion of catecholamines |
Congenital adrenal hyperplasia
This is a group of disorders of adrenal steroid synthesis. Excessive secretion of androgens by the adrenal cortex may occur as a result of a number of conditions of the adrenal gland. However, the most common disorder affecting children is congenital adrenogenital hyperplasia, an inborn deficiency of various enzymes necessary for the biosynthesis of adrenal steroidogenesis (Hockenberry et al 2003). It is an autosomal recessive disorder. Interference with the biosynthesis of glucocorticoids in fetal life results in cortisol secretion being diminished and an increased production of adrenocorticotrophic hormone, which stimulates hyperplasia of the adrenal gland. There is an increased production of various adrenal hormone precursors, including androgens. Deficiency of enzymes necessary for biosynthesis of mineralocorticoids may result in diminished secretion of aldosterone.
In its severe form, excess adrenal androgen production beginning in the first trimester of fetal development results in virilisation of the female infant and life-threatening hypovolaemic and hyponatraemic shock (adrenal crisis) in the newborn.
Assessment and diagnosis
Due to the block in the adrenal production of corticosteroids a build up of androgenic precursors occur and this may lead to ambiguous genitalia in the newborn baby. This is rare. In the female with potentially normal ovaries and uterus, virilisation occurs and sexual development is therefore along heterosexual lines. The abnormality of the external genitalia may vary from mild enlargement of the clitoris to complete fusion of the labioscrotal folds, forming a scrotum, a penile urethra, a penile shaft, and enlargement of the clitoris to form a normal-sized gland.
Signs of adrenal insufficiency (salt loss) may be present during the first days of life. This is an acute presentation with hyponatraemia, hyperkalaemia, hypoglycaemia, dehydration, hypotension and circulatory collapse. In the older child, presentation is usually when the enzyme defect is milder, salt loss may not occur – this accounts for approximately 25% of cases (Ball & Bindler 2006). If untreated, growth rate and skeletal maturation are accelerated. Pubic hair appears early, acne may be excessive and voice may deepen. Excessive pigmentation may develop as a result of melanocyte stimulation by excessive production of adrenocorticotrophic hormone (Kappy et al 2003).
In males, sexual development proceeds normally. Male infants may appear normal at birth but present with salt-losing crisis in the first 2–4 weeks. The infant may present with vomiting, poor weight gain, poor feeding and electrolyte imbalance. The older male child who was not losing sodium may present with rapid growth and precocious sexual development.
Hormonal studies are essential for accurate diagnosis. Adrenal ultrasonography, CT scanning, and MRI may define pelvic anatomy or enlarged adrenals or indicate the presence of an adrenal tumour. Pelvic ultrasonography may help in delineating the internal anatomy of a newborn with ambiguous genitalia.
Nursing care and treatment
Initially, the recognition of ambiguous genitalia in the newborn requires to be attended to immediately in relation to informing the parents and commencing investigations to confirm sexual identity. Early diagnosis and treatment is crucial. Aims of treatment consist of normalising growth velocity and skeletal maturation using the smallest dose of glucocorticoids that will suppress adrenal function. Mineralocorticoid replacement helps to sustain normal electrolyte homeostasis, although excessive use may result in hypertension.
Patient education is necessary as has been described for other endocrine disorders. Parents should understand the child’s need for life-long therapy and the side effects, as well as signs of underdosage and requirements during illness. The need for compliance requires to be stressed and the importance of the child carrying appropriate identification. Surgical intervention may be necessary for reconstruction of the female genitalia as soon as possible during infancy in an effort to support ongoing physical and psychological development. Multiple surgical procedures may be necessary, which will require ongoing psychological support for the child and parents. They require ongoing support as they come to terms with the sex of the baby and decisions that they may have to make regarding the need for surgical interventions. Reassurance regarding surgical interventions and the future care and treatment, which will be a life-long commitment, need to be discussed.
Ambiguity in relation to the sex of the newborn can have a devastating effect on parents and other siblings. At this time, anticipation of parental reaction, which may be likened to the process of bereavement and loss, may enhance the nurse’s ability to engage in a more therapeutic way at this time. Parents need to have information in relation to the infant’s condition reinforced while at the same time being provided with the opportunity to express their own sorrow, loss and disappointment. A decision regarding gender should only be made once all investigations are completed and naming the baby should also be delayed until gender determination is made. Their beliefs and values on gender need to be explored. The healthcare professional should avoid referring to the baby as ‘your son’ or ‘your daughter’ and instead refer to the baby as ‘your baby’. Genetic counselling should be provided for the child when they reach adolescence and supportive counselling should be organised for the parents along with genetic screening for any future pregnancies.
As the hereditary form of adrenogenital hyperplasia is an autosomal-recessive disorder, parents should be referred for genetic counselling.
 Activity
Activity
ActivityReview Chapter 44 and Chapter 45:
• How does the knowledge of both chapters relate to the care of the child with diabetes?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Get Clinical Tree app for offline access
