Katherine I. Byar
Care of Patients with Hematologic Problems
Learning Outcomes
Safe and Effective Care Environment
1 Examine individual patient factors for threats to safety, especially among older adults.
2 Apply the principles of asepsis to protect immunocompromised patients.
3 Modify the environment to protect patients who have thrombocytopenia.
6 Verify patient identification before any form of transfusion therapy.
Health Promotion and Maintenance
Psychosocial Integrity
Physiological Integrity
15 Identify three clinical manifestations common to patients who have any type of anemia.
16 Identify people at increased genetic risk for a hematologic disorder.
17 Prioritize nursing care for the patient who has sickle cell disease.
18 Identify the risk factors for the development of leukemia, lymphoma, and myelodysplastic syndromes.
20 Prioritize nursing interventions for the patient with neutropenia.
21 Prioritize nursing interventions for the patient with thrombocytopenia.
22 Prioritize nursing responsibilities during transfusion therapy.
23 Identify patients at risk for complications of transfusion therapy.

http://evolve.elsevier.com/Iggy/
Answer Key for NCLEX Examination Challenges and Decision-Making Challenges
Audio Glossary
Concept Map Creator
Key Points
Review Questions for the NCLEX® Examination
Hematologic problems result from the impaired production, the impaired function, or the abnormal destruction of any type of blood cell. Problems of the hematologic system can affect many tissues and organs by interfering with oxygenation and tissue perfusion. The type and severity of the disorder determine the impact it has on the health of patients. This chapter discusses mild hematologic disorders and those that are potentially life threatening, such as sickle cell disease and leukemia.
Red Blood Cell Disorders
Red blood cells (RBCs), also known as erythrocytes, are the major cell in the blood. Tissue oxygenation depends on keeping the circulating number of RBCs within the normal range for the person’s age and gender. It also depends on the ability of RBCs to perform their normal functions. RBC disorders include problems in production, function, and destruction. Problems may result in poor function of RBCs, decreased numbers of RBCs (anemia), or an excess of RBCs (polycythemia).
Anemia
Anemia is a reduction in either the number of RBCs, the amount of hemoglobin, or the hematocrit (percentage of packed RBCs per deciliter of blood). It is a clinical sign, not a specific disease, because it occurs with many health problems. Anemia can result from dietary problems, genetic disorders, bone marrow disease, or excessive bleeding. GI bleeding is the most common reason for anemia in adults.
There are many types and causes of anemia. Some are caused by a deficiency in one or more of the components needed to make fully functional RBCs. Such anemias can be caused by deficiencies of iron, vitamin B12, folic acid, or intrinsic factor. Other causes include a decreased rate of RBC production and increased RBC destruction. Table 42-1 lists common causes of many anemias. Despite the many causes of anemia, the effects of anemia on the patient (Chart 42-1) and the nursing care needed are similar for all types of anemia (Coyer & Lash, 2008).
TABLE 42-1
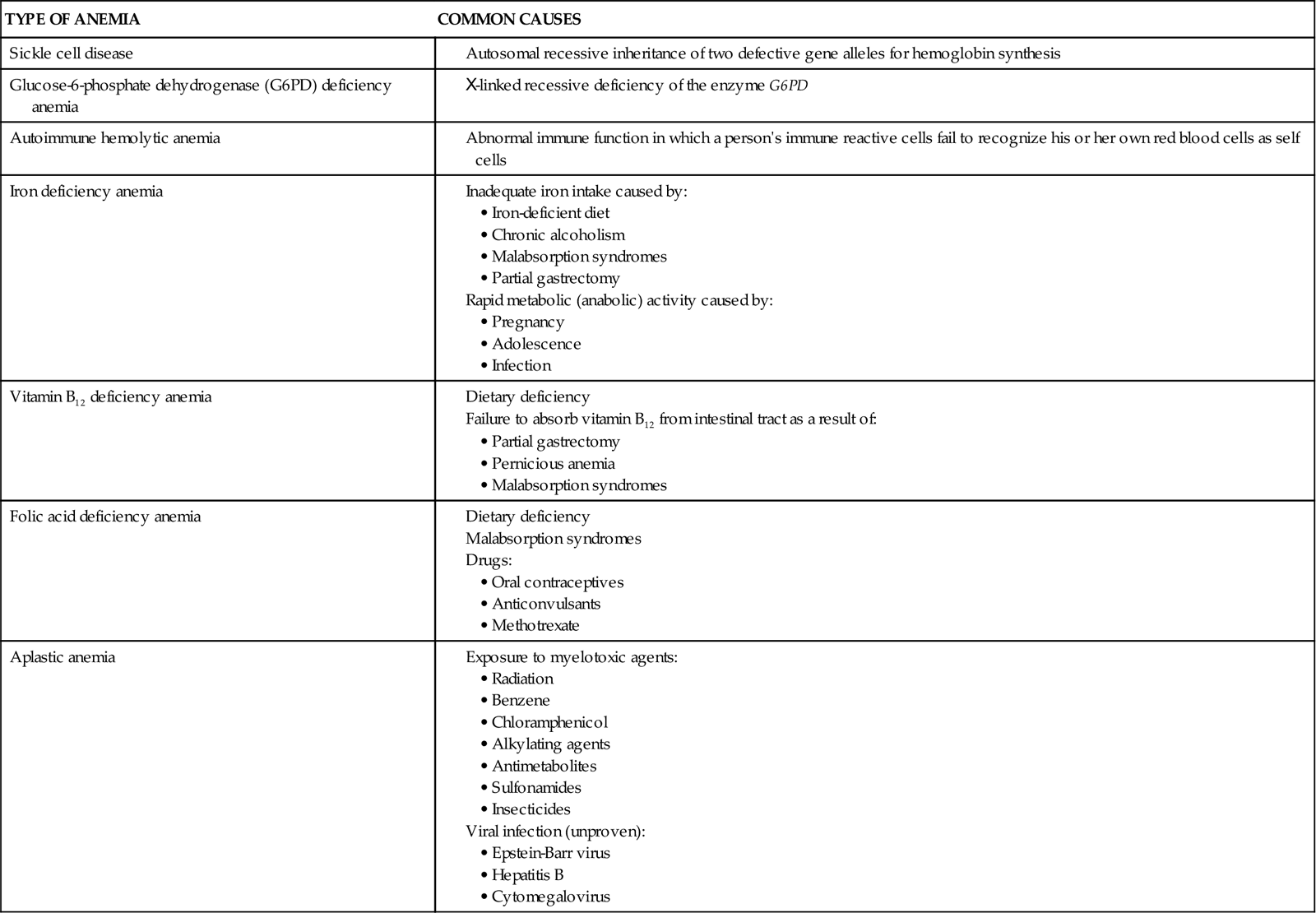
Anemias Resulting From Increased Destruction of Red Blood Cells
Sickle Cell Disease
Pathophysiology
Sickle cell disease (SCD), which used to be called sickle cell anemia, is a genetic disorder that results in chronic anemia, pain, disability, organ damage, increased risk for infection, and early death. There is a difference between sickle cell disease state and sickle cell trait. In addition, there is great variation among patients in how severe the disease is and when complications start.
The main problem in this disorder is the formation of abnormal hemoglobin chains. In healthy adults, the normal hemoglobin molecule has two alpha chains and two beta chains of amino acids. Normal adult hemoglobin is called hemoglobin A (HbA). Normal adult red blood cells usually contain 98% to 99% HbA, with a small percentage of a fetal form of hemoglobin (HbF).
In SCD, at least 40% (and often much more) of the total hemoglobin is composed of an abnormality of the beta chains, known as hemoglobin S (HbS). HbS is sensitive to changes in the oxygen content of the RBC. When RBCs having large amounts of HbS are exposed to decreased oxygen conditions, the abnormal beta chains contract and pile together within the cell, distorting the shape of the RBC. These cells assume a sickle shape, become rigid, and clump together, causing the RBCs to become “sticky” and fragile. These clumps form masses of sickled RBCs that block blood flow (Fig. 42-1). This blood vessel obstruction, known as a vaso-occlusive event (VOE), leads to further tissue hypoxia (reduced oxygen supply) and more sickle-shaped cells, which then leads to more blood vessel obstruction and ischemia. Repeated episodes of ischemia lead to progressive organ damage from anoxia and infarction. Conditions that cause sickling include hypoxia, dehydration, infections, venous stasis, pregnancy, alcohol consumption, high altitudes, low or high environmental or body temperatures, acidosis, strenuous exercise, emotional stress, and anesthesia.
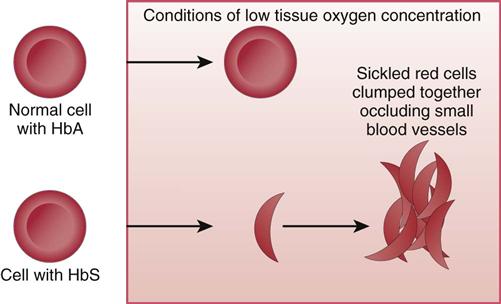
Usually, sickled cells go back to normal shape when the precipitating condition is removed, the blood oxygen level is normalized, and proper tissue perfusion resumes. Although the cells then appear normal, at least some of the hemoglobin remains twisted, decreasing cell flexibility. The cell membranes become damaged over time, and cells are permanently sickled. The membranes of cells with HbS are more fragile and more easily broken. The average life span of an RBC containing 40% or more of HbS is about 12 to15 days, much less than the 120-day life span of normal RBCs. This reduced RBC life span causes hemolytic (blood cell–destroying) anemia in patients with sickle cell disease.
The patient with SCD has periodic episodes of extensive cellular sickling, called crises. The crises have a sudden onset and can occur as often as weekly or as seldom as once a year. Many patients are in good health much of the time, with crises occurring only in response to conditions that cause local or systemic hypoxemia (deficient oxygen in the blood).
Repeated VOEs of larger blood vessels have long-term damaging effects on tissues and organs. Most effects occur as a result of tissue hypoxia, anoxia, ischemia, and cell death. Tissues and organs begin to have small infarcted areas and scar tissue formation. Eventually, so many healthy cells are destroyed that organ failure results. Tissues most often affected in this way are the spleen, liver, heart, kidney, brain, joints, bones, and retina.
Etiology and Genetic Risk
Sickle cell disease (SCD) is a genetic disorder with an autosomal recessive pattern of inheritance (see Chapter 6). The formation of the beta chains of hemoglobin depends on a pair of gene alleles (alternate forms of a gene). A mutation in these alleles leads to the formation of HbS instead of HbA. In sickle cell disease, the patient has two HbS gene alleles, one inherited from each parent, usually resulting in 80% to 100% of the total hemoglobin being HbS. Because both hemoglobin alleles are S, sickle cell disease is sometimes also abbreviated “SS.” Patients with SCD have severe manifestations of the disease even when triggering conditions are mild. If a patient with SCD has children, each child will inherit one of the two abnormal gene alleles and at least have sickle cell trait.
In sickle cell trait, one normal gene allele and one abnormal gene allele for hemoglobin are inherited, so that only half of the hemoglobin chains produced are abnormal. Because people with this problem have one hemoglobin A allele and one hemoglobin S allele, sickle cell trait is abbreviated “AS.” The patient is a carrier of the HbS gene allele (Fig. 42-2) and can pass the trait on to his or her children. However, the patient has only mild manifestations of the disease when precipitating conditions are present because less than 40% of the hemoglobin is abnormal.
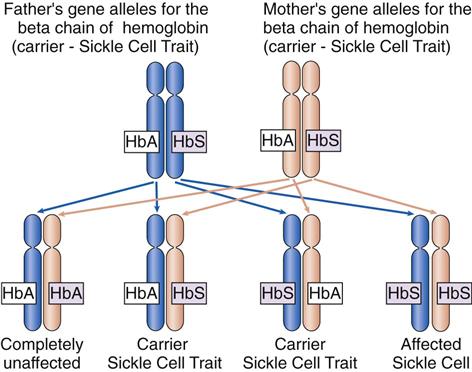
Incidence/Prevalence
Sickle cell trait and different forms of SCD occur in people of all races and ethnicities but less often among white people. In the United States, about 70,000 people have SCD, most commonly in African Americans (U.S. National Library of Medicine [USNLM], 2011). SCD occurs in 1 in 500 African Americans, and 1 in 12 to 1 in 15 (8%) African Americans are carriers of one sickle cell gene allele and have AS (USNLM, 2011).
Patient-Centered Collaborative Care
Assessment
History
An adult with sickle cell disease (SCD) usually has a long-standing diagnosis of the disorder. An adult with sickle cell trait usually has no hematologic manifestations or abnormal laboratory findings other than the presence of hemoglobin S. This person may be unaware that he or she has a hematologic problem until an acute illness is present or when anesthesia is administered.
Ask the patient about previous crises, what led to the crises, severity, and usual treatments. Explore recent contact with ill people and activities to determine what caused the current crisis. Ask him or her about symptoms of infection as the cause of a crisis, such as sore throat, cough, GI changes, or pain and burning upon urination.
Review all activities and events during the past 24 hours, including food and fluid intake, exposure to temperature extremes, drugs taken, exercise, trauma, stress, recent airplane travel, and ingestion of alcohol or other recreational drugs. Ask about changes in sleep and rest patterns, ability to climb stairs, and any activity that induces shortness of breath. Determine his or her perceived energy level using a scale ranging from 0 to 10 (0 = not tired with plenty of energy; 10 = total exhaustion) to assess the degree of fatigue. This activity review provides important information about fatigue, activity tolerance, and participation in ADLs. It may also help patients determine what specific types of activities or environmental changes trigger their crises.
Physical Assessment/Clinical Manifestations
Pain is the most common symptom of SCD crisis. Other manifestations vary with the site of tissue damage.
Cardiovascular changes, including the risk for high-output heart failure, occur because of the anemia. Assess the patient for shortness of breath and general fatigue or weakness. Other problems may include murmurs, the presence of an S3 heart sound, and increased jugular-venous pulsation or distention. Assess his or her cardiac and vascular status by comparing peripheral pulses, temperature, and capillary refill in all extremities. Extremities distal to blood vessel occlusion are cool to the touch with slow capillary refill and may have reduced or absent pulses. Heart rate may be rapid and blood pressure low to average, with a decreased pulse pressure, because breakage of RBCs leads to anemia. Anemia results in a less viscous (thinner) blood that moves more rapidly through the heart and blood vessels under lower pressures than does blood with normal viscosity.
Priapism is a prolonged penile erection that can occur in men who have SCD. The cause is excessive vascular engorgement in erectile tissue. The condition is very painful and can last for hours. During the priapism episode, the patient usually cannot urinate.
Skin changes include pallor or cyanosis because of poor oxygenation from decreased perfusion and anemia. Examine the lips, tongue, nail beds, conjunctivae, palms, and soles of the feet at least every 8 hours for subtle color changes. With cyanosis, the lips and tongue are gray and the palms, soles, conjunctivae, and nail beds have a bluish tinge.
Another skin manifestation of SCD is jaundice. Jaundice results from RBC destruction and release of bilirubin. Bilirubin is present inside RBCs and released when fragile cells are damaged, leading to jaundice. To assess for jaundice in patients with darker skin, inspect the roof of the mouth for a yellow appearance. Yellow-tinged sclera may be misleading because of normal deposits of fat that appears yellowish in contrast to the dark skin around the eye. Examine the sclera closest to the cornea to assess jaundice more accurately. The palms and soles of dark-skinned patients may appear yellow if calloused and could be mistaken for jaundice. Jaundice often causes intense itching.
Despite the anemia, patients with SCD usually are not iron deficient. With increased RBC production and destruction, iron released from the cells increases the pigmentation of the skin.
As many as 75% of adults with SCD have open sores or ulcers on the lower legs that are caused by poor tissue perfusion. The outer sides and inner aspect of the ankle or the shin are common ulcer sites. Inspect the legs and feet for open lesions or darkened areas that may indicate necrotic tissue. These lesions often become necrotic or infected, requiring débridement and antibiotic therapy. Skin grafting may be needed when ulcers fail to heal.
Abdominal changes include major organ damage to the spleen and liver. These organs are usually the first to be damaged from many episodes of hypoxia and ischemia. In crisis, abdominal pain is diffuse and steady, involving the back and legs (Sickle Cell Information Center, 2010). Bowel sounds should be present with normal activity. Palpate the liver and spleen for enlargement. The liver or spleen may feel firm and enlarged with a nodular or “lumpy” texture in later stages of the disease. Check for guarding or rebound tenderness to palpation. A rapidly enlarging liver or spleen with increasing jaundice may indicate blood trapping in those organs.
Kidney and urinary changes are common as a result of poor perfusion. Chronic kidney disease occurs as a result of anoxic damage to the kidney nephrons. With early damage, the kidneys are less effective at filtration and reabsorption. The urine contains protein, and the patient may not concentrate urine. Eventually, the kidneys fail, resulting in little or no urine output. The buildup of waste products in the blood leads to death.
Musculoskeletal changes occur because arms and legs are common sites of blood vessel occlusion in SCD. Joints also may be damaged from hypoxic episodes and have necrotic degeneration. Inspect the arms and legs for symmetry, and record any areas of swelling, temperature, or color difference. Ask patients to move all joints. Record the range of motion and any pain with movement.
Central nervous system (CNS) changes may occur in SCD. During crises, patients may have a low-grade fever. If the CNS has infarcts or repeated episodes of hypoxia, patients may have seizures or manifestations of a stroke, including pronator drift and a weakened hand grasp. Assess for the presence of “pronator drift” by having the patient extend both arms in front in a palms up (supination) position. Pronator drift occurs when the patient cannot maintain one or both hands in the supine position and the hand drifts into a prone or palm-down position. Assess hand grasps on both sides. Also assess the patient’s gait and coordination.
Psychosocial Assessment
Psychosocial assessment is important because behavioral changes are early manifestations of cerebral hypoxia from poor tissue perfusion. Observe the patient, and document behavior. Ask family members whether the current behavior and mental status are usual for the patient.
SCD is a painful, life-limiting disorder that can be passed on to one’s children. Assess the patient’s psychosocial needs in terms of new factors, established support systems, use of coping patterns, and disease progression. Ask the patient about how he or she views the disease and what changes in lifestyle have been made as limitations increased.
Laboratory Assessment
The diagnosis of SCD is based on the percentage of hemoglobin S (HbS) on electrophoresis. A person who has AS usually has less than 40% HbS, and the patient with SCD may have 80% to 100% HbS. This percentage does not change during crises. Another indicator of SCD is the number of RBCs with permanent sickling. This value is less than 1% among people with no hemoglobin disease, is 5% to 50% among people with AS, and may be 90% among patients with SCD.
Other laboratory tests can indicate complications of the disease, especially during crises. The hematocrit of patients with SCD is low (between 20% and 30%) because the life span of the RBC is so much shorter than normal and because many cells are destroyed. This value decreases even more during crises or when the bone marrow fails to produce cells during stress (aplastic crisis). The reticulocyte count is high, indicating anemia of long duration. It is high because the bone marrow is releasing immature red blood cells (reticulocytes) to make up for the low hematocrit and resulting tissue hypoxia. Often the total bilirubin levels are high in patients with SCD because the hemoglobin released from damaged and dead red blood cells is broken down into iron and heme molecules. The heme molecules then are metabolized into free bilirubin.
The total white blood cell (WBC) count is usually high in patients with SCD. This elevation is related to chronic inflammation caused by tissue hypoxia and ischemia.
Imaging Assessment
Bone changes occur as a result of chronically stimulated marrow and low bone oxygen levels. The skull may show changes on x-ray as a result of bone surface cell destruction and new growth, giving the skull a “crew cut” appearance on x-ray. X-rays of joints may show necrosis and destruction. Ultrasonography, computed tomography (CT), positron emission tomography (PET), and magnetic resonance imaging (MRI) may show soft-tissue and organ changes from poor oxygenation and chronic inflammation.
Other Diagnostic Assessment
Electrocardiographic (ECG) changes document cardiac infarcts and tissue damage. Specific ECG changes are related to the area of the heart that has been damaged. Echocardiograms may show cardiomyopathy and decreased cardiac output (low ejection fraction).
 NCLEX Examination Challenge
NCLEX Examination ChallengeAnalysis
The priority problems for the patient with sickle cell disease are:
Interventions
Managing Pain
The most common problem of SCD is pain (Granados & Jacob, 2009). The pain with sickle cell crisis is the result of tissue injury caused by poor oxygenation from obstructed blood flow. At times, patients have mild pain episodes that can be managed at home. However, pain is often severe enough to require hospitalization and large doses of opioid analgesics. Acute pain episodes have a sudden onset, usually involving the chest, back, abdomen, and extremities. Complications of SCD, such as bone necrosis, can cause severe, chronic pain, requiring large doses of opioid analgesics.
Ask the patient whether the pain is typical of past pain episodes. If not, other pain causes or disease complications must be explored. Use of a pain rating scale can help proper pain management. Ask the patient to rate pain on a scale ranging from 0 to 10, and evaluate the effectiveness of interventions based on the ratings.
Concerns about substance abuse can lead to inadequate pain treatment in these patients. Opioid addiction is rare, occurring in only 2% to 5% of patients with SCD. Pain management is based on past pain history, previous drug use, disease complications, and current pain assessment. Health care providers need to be aware of their own attitudes when caring for this population. If substance abuse is suspected, management of addiction is incorporated into the patient’s overall treatment plan. Management of substance abuse and SCD poses many challenges, because the patient usually cannot be expected to be totally opioid-free. Addicted patients with acute pain crisis may need opioids for short periods.
Drug therapy for patients in acute sickle cell crisis often starts with at least 48 hours of IV analgesics. (Chart 42-2 lists best practices for nursing care of the patient in sickle cell crisis.) Morphine and hydromorphone (Dilaudid) are given IV on a routine schedule or by infusion pump using patient-controlled analgesia (PCA) (see Chapter 5). Once relief is obtained, the IV dose can be tapered and the drug given orally. Avoid “as needed” (PRN) schedules because they do not provide adequate relief. Avoid IM injections because absorption is impaired by poor perfusion and sclerosed skin. Moderate pain may be treated with oral opioids or NSAIDs. (See Chapter 5 for more information on pain management.)
Hydroxyurea (Droxia) has been successfully used to reduce the number of sickling and pain episodes (Pack-Mabien & Haynes, 2009). Hydroxyurea works by stimulating fetal hemoglobin (HbF) production. HbF is present during fetal development, but its production is turned off before birth. Increasing the level of HbF reduces sickling of red blood cells in patients with sickle cell disease. However, this drug is associated with an increased incidence of leukemia. Long-term complications should be discussed with the patient before this therapy is started. Hydroxyurea also suppresses bone marrow function, and regular follow-up to monitor complete blood counts (CBCs) for drug toxicity is important. Hydroxyurea also causes birth defects and should not be taken by anyone who is pregnant or likely to become pregnant.
 Nursing Safety Priority
Nursing Safety Priority
Action Alert
Teach sexually active women of childbearing age to use at least two methods of birth control while taking hydroxyurea and for 1 month after the drug is discontinued because this drug can cause severe birth defects.
Hydration by the oral or IV route helps reduce the duration of pain episodes. Urge the patient to drink water or juices. Hypotonic fluids, such as dextrose 5% in water (D5W) or dextrose 5% in 0.45% sodium chloride, are usually infused at 250 mL/hr for 4 hours. Hypotonic fluids are used because the patient’s blood volume is usually hypertonic as a result of dehydration. Using hypotonic fluids can help bring the patient’s blood osmolarity back down to the normal range of 270 to 300 mOsm. The IV rate is then reduced to 125 mL/hr if more hydration is needed.
Complementary therapies and other measures, such as keeping the room warm, using distraction and relaxation techniques, positioning with support for painful areas, aroma therapy, therapeutic touch, and warm soaks or compresses, all help reduce pain perception.
 Nursing Safety Priority
Nursing Safety Priority
Action Alert
Do not assume that complementary therapies alone will provide adequate pain relief. Analgesics are needed to manage sickle cell pain.
Preventing Sepsis
The patient with SCD is at greater risk for bacterial infection because of decreased spleen function resulting from anoxic damage to the spleen. Over time, the spleen may become completely nonfunctional. Expected outcomes of interventions include preventing infection, controlling infection, and starting treatment early for specific infections. The patient who develops a fever should have diagnostic testing for sepsis including CBC with differential, blood cultures, reticulocyte count, urine culture, and a chest x-ray. Usually these patients are started on prophylactic antibiotics.
Prevention and early detection strategies are used to protect the patient in sickle cell crisis from infection. Frequent, thorough handwashing is of the utmost importance. Any person with an upper respiratory tract infection who must enter the patient’s room wears a mask. Use strict aseptic technique for all invasive procedures. Coordinate with all members of the health care team to ensure adherence to these strategies.
Continually assess the patient for infection, and monitor the daily CBC with differential WBC count. Inspect the mouth every 8 hours for lesions indicating fungal or viral infection. Listen to the lungs every 8 hours for crackles, wheezes, or reduced breath sounds. Each time the patient voids, inspect the urine for odor and cloudiness, and ask about urgency, burning, or pain during urination. Take vital signs at least every 4 hours to assess for fever, or supervise this action when performed by others.
Drug therapy is a major defense against the infections that develop in the patient with SCD. Prophylactic therapy with twice-daily oral penicillin reduces the number of pneumonia and other streptococcal infections. Urge the patient to receive yearly influenza vaccinations and to receive the pneumonia vaccine. Drug therapy for an actual infection can control infection and prevent sepsis. Drugs used depend on the sensitivity of the specific organism causing the infection, as well as on the extent of the infection.
Preventing Multiple Organ Dysfunction
Continued blood vessel occlusion by clumping of sickled cells increases the risk for multiple organ dysfunctions. Acute chest syndrome, in which a vaso-occlusive episode (VOE) causes infiltration and damage to the pulmonary system, is a major cause of death in adults with SCD (Hernandez & Patterson, 2009). Preventing dysfunction of any organ is important in SCD management; however, preventing heart and lung damage is a priority. Management of SCD focuses on prevention of VOEs and promotion of oxygenation.
The patient in sickle cell crisis is admitted to the acute care hospital. Assess for adequate perfusion to all body areas. Remove restrictive clothing, and instruct the patient to avoid flexing the knees and hips.
Hydration is needed because dehydration increases cell sickling and must be avoided. Assist him or her in maintaining adequate hydration. The patient in acute crisis needs an oral or IV fluid intake of at least 200 mL/hr.
Oxygen is given during crises because lack of oxygen is the main cause of sickling. Ensure that oxygen therapy is nebulized to prevent dehydration. Monitor oxygen saturation using pulse oximetry. Patients with low oxygen saturation should have an arterial blood gas (ABG) drawn and a chest x-ray.
Transfusion with RBCs can be helpful to increase HbA levels and dilute HbS levels. During transfusion, monitor the patient for complications of the procedure (discussed on pp. 900-901 in the Transfusion Reactions section).
Transfusions are prescribed cautiously to prevent iron overload from repeated transfusions. Iron overload damages the heart, liver, and endocrine organs. Monitor the patient’s serum ferritin, serum iron (Fe), and total iron-binding capacity (TIBC). Deferoxamine mesylate (Desferal, Desferrioxamine) or deferasirox (Exjade) may be prescribed to manage transfusion-induced iron overload.
In some treatment centers, hematopoietic stem cell transplantation (HSCT) is performed to correct abnormal hemoglobin permanently. Because HSCT is expensive and may result in life-threatening complications, its risks and benefits need to be considered for each patient.
 Decision-Making Challenge
Decision-Making Challenge
Safety; Patient-Centered Care
A 23-year-old African-American woman who has a history of sickle cell disease is brought to the emergency department. She reports joint and back pain that is becoming worse rapidly. She rates her pain as a 9 on a 0-to-10 scale. She is pale, and her vital signs are normal except for a temperature of 101.5° F (38.6° C). She tells you that she had sexual intercourse for the first time 2 days ago. She also says that she now has pain just above the pubic bone during and after urination and that the urine burns.
1 What questions related to pain would you ask this patient?
2 What drug or drugs for pain would you expect this patient to receive and by which route?
3 What diagnostic tests would you expect to be requested for this patient?
4 What nursing assessments would be most important for you to make at this time?
5 What factor or factors could have triggered this crisis episode?
Community-Based Care
Sickle cell disease (SCD) becomes worse over time. Rarely is there a true remission, although the number of crisis episodes may be reduced. Care focuses on teaching the patient and family how to prevent crises and complications (Chart 42-3). The patient with SCD may receive care in acute care, subacute care, extended or assistive care, and home care settings.
Teach the patient to avoid specific activities that lead to hypoxia and hypoxemia. Stress the recognition of the early symptoms of crisis so that interventions can be started early to prevent pain, complications, and permanent tissue damage. He or she is often given opioid analgesics for self-management of sickle cell crises at home. Teach the patient and family about the correct use of these drugs. In addition, counsel patients about the hereditary aspects of SCD and provide information about prenatal diagnosis, birth control methods, and pregnancy options.
 NCLEX Examination Challenge
NCLEX Examination Challenge
Psychosocial Integrity
A woman whose hemoglobin S levels are less than 1% has a brother with sickle cell disease (SCD) and both parents have been diagnosed as carriers for the disorder. She asks what her risks are of having a child with sickle cell disease. What is the nurse’s best response?
Glucose-6-Phosphate Dehydrogenase Deficiency Anemia
Pathophysiology
Many forms of hemolytic (blood cell–destroying) anemia are present from birth as a result of defects or deficiencies of one or more enzymes in red blood cells (RBCs). More than 200 such disorders are known. Most of these enzymes are needed to complete some critical step in RBC energy production. The most common type of inherited hemolytic anemia is the deficiency of the enzyme glucose-6-phosphate dehydrogenase (G6PD). This disease is inherited as an X-linked recessive disorder, most fully expressed in homozygous males, although partial expression (carrier state) is possible in heterozygous females. This type of anemia affects about 10% of all African Americans and also may occur in Sephardic Jews, Greeks, Iranians, Chinese, Filipinos, and Indonesians, with a frequency ranging from 5% to 40% (McCance et al., 2010).
G6PD stimulates reactions in glucose metabolism. Because RBCs contain no mitochondria (sites of production of adenosine triphosphate [ATP]), metabolism of glucose is required to generate energy in these cells. Newly produced RBCs from patients with G6PD deficiency have sufficient levels of G6PD. Cells with reduced amounts of G6PD break more easily during exposure to some drugs (e.g., sulfonamides, aspirin, quinine derivatives, rasburicase, chloramphenicol, dapsone, high doses of vitamin C, and thiazide diuretics) and exposure to benzene and other toxins.
The patient usually does not have symptoms until exposed to these agents or until a severe infection develops. After exposure to any of these agents, patients have acute breakage of RBCs lasting from 7 to 12 days. During this acute phase, anemia and jaundice develop. The hemolytic reaction is limited because only older RBCs, containing less G6PD, are destroyed.
Patient-Centered Collaborative Care
Prevention is the most important therapeutic measure. Men who belong to the high-risk groups should be tested for this problem before being given drugs that can cause the hemolytic reaction. Screening test results are based on discoloration of methylene blue and reduction of methemoglobin. Donated blood is screened for this deficiency before transfusion because cells deficient in G6PD can be hazardous for the recipient.
Hydration is important during an episode of hemolysis to prevent debris and hemoglobin from collecting in the kidney tubules, which can lead to acute kidney failure. Osmotic diuretics, such as mannitol (Osmitrol), may help prevent this complication. Transfusions are needed when anemia is present and kidney function is normal (see Transfusion Therapy section on pp. 897-901).
Immunohemolytic Anemia
The most common types of hemolytic anemias in North America are the immunohemolytic anemias, also referred to as autoimmune hemolytic anemias (McCance et al., 2010). The pathophysiology of hemolytic anemias involves the excessive destruction of red blood cells followed by acceleration of erythropoiesis. Acquired hemolytic syndromes result from increased RBC destruction occurring in response to trauma, viral infection, malarial infection, exposure to certain chemicals or drugs, and autoimmune reactions. All increase the rate of RBC destruction by causing membrane lysis (breakage).
In immunohemolytic anemia, immune system products (e.g., antibodies) attack a person’s own RBCs for unknown reasons. Some hemolytic anemias occur with other autoimmune disorders (e.g., systemic lupus erythematosus) or other conditions such as lymphoma, leukemia, and other neoplastic disorders (Coyer & Lash, 2008). Regardless of the cause, RBCs are viewed as non-self by the immune system and then are attacked and destroyed.
The two types of immunohemolytic anemia are warm antibody anemia and cold antibody anemia. Warm antibody anemia occurs with immunoglobulin G (IgG) antibody excess. These antibodies are most active at 98.6° F (37° C) and may be triggered by drugs, chemicals, or other autoimmune problems. Cold antibody anemia has complement protein fixation on immunoglobulin M (IgM) and occurs most at 86° F (30° C). This problem often occurs with a Raynaud’s-like response in which the arteries in the hands and feet constrict profoundly in response to cold temperatures or stress.
Management depends on symptom severity. Steroid therapy to suppress immune function is the first line of treatment and is temporarily effective in most patients. Splenectomy and more intense immunosuppressive therapy with cancer chemotherapy drugs may be used if steroid therapy fails. Plasma exchange therapy to remove attacking antibodies is effective for patients who do not respond to immunosuppressive therapy.
Anemias Resulting From Decreased Production of Red Blood Cells
Anemias caused by decreased RBC production occur in response to many problems. Some are caused by failure of the bone marrow to produce healthy RBCs. Anemias also are caused by failure of the body to make or absorb a substance needed for RBC production.
Iron Deficiency Anemia
Adults usually have between 2 and 6 g of iron, depending on the size of the person and the amount of hemoglobin in the cells. About two thirds of this iron is contained in hemoglobin. The other one third is stored in the bone marrow, spleen, liver, and muscle. With iron deficiency, the iron stores are depleted first, followed by the hemoglobin stores. As a result, RBCs are small (microcytic) and the patient has mild symptoms of anemia, including weakness and pallor. Other clinical manifestations include fatigue, reduced exercise tolerance, and fissures at the corners of the mouth. Nails become brittle, thin, coarsely ridged, or spoon-shaped and concave (Coyer & Lash, 2008). In iron deficiency anemia, serum ferritin values are less than 10 ng/mL (normal range is 12 to 300 ng/mL).
Iron deficiency anemia is the most common anemia worldwide, especially among women, older adults, and people with poor diets. It can result from blood loss, poor GI absorption of iron, and an inadequate diet (Simmons, 2010). The problem is a decreased iron supply for the developing RBC.
Any adult with iron deficiency should be evaluated for abnormal bleeding, especially from the GI tract. Management of iron deficiency anemia involves increasing the oral intake of iron from food sources (e.g., red meat, organ meat, egg yolks, kidney beans, leafy green vegetables, and raisins). An adequate diet supplies about 10 to 15 mg of iron per day. However, only 5% to 10% of dietary iron is absorbed. This amount is enough to meet the needs of men and of women after childbearing age but is not sufficient to supply the greater needs of menstruating women. If iron losses are mild, oral iron supplements, such as ferrous sulfate, are started. This treatment should cause the hemoglobin level to rise about 2 g/dL in 4 weeks. Treatment continues until the hemoglobin level returns to normal. Instruct patients to take the iron supplement between meals for better absorption and to reduce GI distress. When iron deficiency anemia is severe, iron solutions can be given IV or IM. When given IM, these solutions must be given using the Z-track best practice method outlined in Chart 42-4. There is controversy as to whether the dorsal gluteal site or the ventrogluteal site should be used for iron dextran (Dexferrum) therapy. Although the ventrogluteal site is recommended as best practice for most IM injections, including those administered by Z-track (Zimmermann, 2010), the drug manufacturer continues to recommend the dorsal gluteal site (MD Consult, 2010). A new IV drug for iron deficiency anemia associated with chronic kidney disease is ferumoxytol (Feraheme). It has the advantage of being administered at higher dosages over a shorter period of time compared with other IV iron preparations (Belavic, 2010).
Vitamin B12 Deficiency Anemia
Production of RBCs depends on adequate DNA synthesis in the precursor cells so that cell division and growth into functional RBCs can occur. All cell division requires adequate amounts of folic acid to make DNA. One function of vitamin B12 is to activate the enzymes that move folic acid into the cell where DNA synthesis occurs. Vitamin B12 deficiency causes anemia by inhibiting folic acid transport and reducing DNA synthesis in precursor cells. These precursor cells then undergo improper DNA synthesis and increase in size. Only a few are released from the bone marrow. This type of anemia is called megaloblastic or macrocytic anemia because of the large size of these abnormal cells.
Vitamin B12 deficiency results from poor intake of foods containing vitamin B12. This more often occurs with vegetarian diets or diets lacking dairy products. Problems such as small bowel resection, chronic diarrhea, diverticula, tapeworm, or overgrowth of intestinal bacteria can lead to poor absorption of vitamin B12. Anemia resulting from failure to absorb vitamin B12 (pernicious anemia) is caused by a deficiency of intrinsic factor (a substance normally secreted by the gastric mucosa), which is needed for intestinal absorption of vitamin B12.
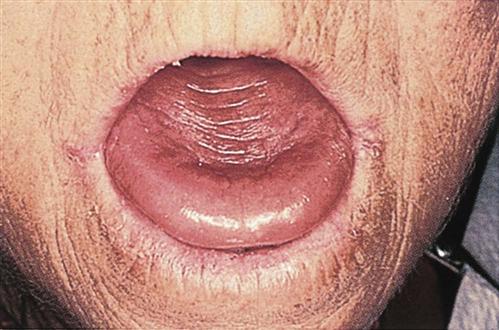
Vitamin B12 deficiency anemia may be mild or severe, usually develops slowly, and produces few symptoms. Patients usually have pallor and jaundice, as well as glossitis (a smooth, beefy-red tongue) (Fig. 42-3), fatigue, and weight loss. Because vitamin B12 is needed for normal nerve function, patients with pernicious anemia may also have paresthesias (abnormal sensations) in the feet and hands and poor balance (Nettina, 2009).
When anemia is caused by a dietary deficiency, the focus of management is to increase the intake of foods rich in vitamin B12 (animal proteins, eggs, and dairy products). Vitamin supplements may be prescribed when anemia is severe. Vitamin B12 and folic acid levels are monitored. Patients who may have pernicious anemia are tested using the Shilling test, which measures the presence of vitamin B12 in the urine after the patient is given an oral dose of radioactive vitamin B12. If the patient does not absorb the radioactive vitamin B12 because of pernicious anemia, it cannot get into the urine. Patients who have pernicious anemia are given vitamin B12 injections weekly at first, then monthly for the rest of their lives. A new formulation of the drug cyanocobalamin (CaloMist) that delivers vitamin B12 by nasal spray also is available to maintain vitamin levels after the patient’s deficiency has first been corrected by the traditional injection method.
Folic Acid Deficiency Anemia
Folic acid deficiency can also cause anemia. Manifestations are similar to those of vitamin B12 deficiency, but nervous system functions remain normal because folic acid deficiency does not affect nerve function. The absence of neurologic problems helps distinguish folic acid deficiency from vitamin B12 deficiency. The disease develops slowly, and symptoms may be attributed to other problems or diseases.
Three causes of folic acid deficiency are poor nutrition, malabsorption, and drugs. Poor nutrition, especially a diet lacking green leafy vegetables, liver, yeast, dried beans, and nuts, is the most common cause. Malabsorption syndromes, such as Crohn’s disease, are the second most common cause. Chronic alcohol abuse with malnutrition is another cause. Anticonvulsants and oral contraceptives can slow or prevent the absorption and conversion of folic acid to its active form, leading to folic acid deficiency and anemia.
Prevention begins by identifying high-risk patients, such as older, debilitated patients with alcoholism; patients at risk for malnutrition; and those with increased folic acid requirements. A diet rich in foods containing folic acid and vitamin B12 prevents a deficiency. By assessing dietary habits for all patients, you can determine which patients are at risk for diet-induced anemias. This type of anemia is managed with scheduled folic acid replacement therapy.
Aplastic Anemia
Pathophysiology
Aplastic anemia is a deficiency of circulating red blood cells (RBCs) because of failure of the bone marrow to produce these cells. It is caused by an injury to the immature precursor cell for red blood cells, known as the pluripotent stem cell. Although aplastic anemia sometimes occurs alone, it usually occurs with leukopenia (a reduction in white blood cells [WBCs]) and thrombocytopenia (a reduction in platelets). These three problems occur together because the damaged bone marrow loses the ability to produce any of these cells. Pancytopenia (a deficiency of all three cell types) is common in aplastic anemia. Disease onset may be slow or rapid.
The most common type of the disease is acquired aplastic anemia. It can be caused by long-term exposure to toxic agents, drugs (see Table 41-3 in Chapter 41), ionizing radiation, or infection. In about 50% of cases, the cause of aplastic anemia is unknown. The disease may follow viral infection, but it is not known exactly how the bone marrow is damaged. The most common hereditary form of the disease is Fanconi’s anemia.
Patient-Centered Collaborative Care
Assess for symptoms of bone marrow failure and poor oxygenation such as weakness, pallor, and petechiae or ecchymosis. A complete blood count (CBC) shows severe macrocytic anemia, leukopenia and thrombocytopenia. A bone marrow biopsy may show replacement of cell-forming marrow with fat. Infection is common with leukopenia, especially neutropenia.
Blood transfusions are used only when the anemia causes disability or when bleeding is life threatening because of low platelet counts. Unnecessary transfusion increases the chances for developing immune reactions to platelets and shortens the life span of the transfused cell. This therapy is discontinued as soon as the bone marrow begins to produce RBCs.
Allogeneic hematopoietic stem cell transplantation (transplantation of bone marrow or stem cells from a sibling or matched unrelated donor) remains the preferred and most successful method of treatment for aplastic anemia that does not respond to other therapies. Cost, availability, and complications limit this treatment in aplastic anemia. For those patients who are unable to undergo such treatment or lack a suitable donor, immunosuppressive therapy remains the treatment of choice (Afable & Lyon, 2008).
Immunosuppressive therapy helps patients who have the types of aplastic anemia with a disease course similar to that of autoimmune problems. Drugs such as prednisone, antithymocyte globulin (ATG), and cyclosporine A (Sandimmune) have resulted in partial or complete remissions. The combination of ATG and cyclosporine A has been found to improve results (Afable & Lyon, 2008). Another immunosuppressant agent that has been found to improve both blood counts and transfusion requirements in patients with moderate aplastic anemia is daclizumab (Zenapax). Splenectomy (removal of the spleen) may be needed for patients with an enlarged spleen that is either destroying normal RBCs or suppressing their development.
 NCLEX Examination Challenge
NCLEX Examination ChallengePolycythemia
In polycythemia, the number of red blood cells (RBCs) in the blood is greater than normal. The blood of a patient with polycythemia is hyperviscous (thicker than normal blood). The problem may be temporary (occurring as a result of other conditions) or chronic. One type of polycythemia, polycythemia vera (PV), is fatal if left untreated.
Polycythemia Vera
Pathophysiology
Polycythemia vera (PV) is a disease with a sustained increase in blood hemoglobin levels to 18 g/dL, an RBC count of 6 million/mm3, or a hematocrit of 55% or greater. PV is a cancer of the RBCs with three major hallmarks: massive production of RBCs, excessive leukocyte production, and excessive production of platelets. Extreme hypercellularity (cell excess) of the peripheral blood occurs in people with PV.
The patient’s facial skin and mucous membranes have a dark, flushed (plethoric) appearance. These areas appear purplish or cyanotic because the blood in these tissues is poorly oxygenated. Most patients have intense itching caused by dilated blood vessels and poor tissue oxygenation. Blood viscosity is increased, resulting in hypertension. Superficial veins are visibly distended. The thick blood moves more slowly through all tissues and places increased demands on the heart, resulting in hypertension. In some highly vascular areas, blood flow may become so slow that stasis occurs. Vascular stasis causes thrombosis (clot formation) within the smaller vessels, occluding blood vessels. The blood vessel occlusion leads to tissue hypoxia, anoxia and, later, to infarction and necrosis. Tissues most at risk for this problem are the heart, spleen, and kidneys, although infarction and damage can occur in any organ or tissue.
Because the actual number of cells in the blood is greatly increased and the cells are not completely normal, cell life spans are shorter. The shorter life spans and increased cell production cause a rapid turnover of circulating blood cells. This rapid turnover increases the amount of cell debris (released when cells die) in the blood, adding to the general “sludging” of the blood. This debris includes uric acid and potassium, which cause the symptoms of gout and hyperkalemia (elevated serum potassium level).
Other manifestations of PV are related to abnormal RBCs. Even though the number of RBCs is greatly increased, their oxygen-carrying capacity is impaired, and patients have poor oxygenation with severe hypoxia. Patients with PV also have bleeding problems because of platelet impairment.
Patient-Centered Collaborative Care
Polycythemia vera is a malignant disease that progresses in severity over time. If left untreated, few people with PV live longer than 2 years after diagnosis. With management by repeated pheresis (two to five times per week), the patient may live 10 to 15 years or longer. (Pheresis is the withdrawal of whole blood and removal of the patient’s RBCs to decrease the number of RBCs and reduce blood viscosity. The remaining plasma is then re-infused back into the patient.) Increasing hydration and promoting venous return help prevent clot formation. The purpose of therapy is to prevent clot formation and includes the use of anticoagulants. Chart 42-5 lists health tips for patients with PV.
As the disease progresses, drug therapy is used to control it, although aggressive IV chemotherapy is no longer recommended because of its increased risk for inducing leukemia. Aspirin therapy may be used to decrease clot formation. However, the benefits must be weighed against the increased risk for GI bleeding associated with this therapy. Hydroxyurea, an oral chemotherapy drug, may be prescribed for severe manifestations of the disease.
Myelodysplastic Syndromes
Pathophysiology
Myelodysplastic syndromes (MDSs) are a group of disorders caused by the formation of abnormal bone marrow cells. These abnormal cells are usually destroyed shortly after they are released into the blood. As a result, patients with MDS have a decrease in all blood cell types. Anemia is a common problem, occurring in 90% of patients with MDS, although neutropenia (low white blood cell [WBC]) count and thrombocytopenia (low platelets) are also often present.
Although MDS can occur at any age, it is most common in people age 60 years or older. MDS is not officially a type of cancer but does have cancer-like features and is considered to be a precancerous state. Like cancer, MDS arises from a single population of abnormal cells. About 30% of all patients with MDS do eventually develop acute leukemia (Kumar, Abbas, Fausto, & Aster, 2010). A number of subtypes of MDS have different prognoses and responses to therapy. Patients are categorized into risk groups (i.e., low, intermediate [1 and 2], high) based on the severity of pancytopenia (low counts of all blood cell types), cytogenetic abnormalities, and numbers of blast cells (immature WBC cells) found in the bone marrow (Kurtin & Demakos, 2010).
The exact cause of MDS is not clear. Risk factors include normal physiologic changes associated with aging, chemical exposures (pesticides, benzene), tobacco smoke, and exposure to radiation or chemotherapy drugs. Diagnosis is made by examination of the chromosomes and the genes within the chromosomes (cytogenetic testing) of the bone marrow cells. Peripheral blood smears are used to assess the level of cell maturation and the proportion of abnormal cells.
Patient-Centered Collaborative Care
The only curative treatment for MDS is an allogeneic hematopoietic stem cell transplantation, which is often not an option because of the advanced age of the patient (Kurtin & Demakos, 2010). For low-risk and intermediate-1–risk MDS, the antitumor immunomodulatory agent lenalidomide (Revlimid) is approved for patients whose dysplastic cells have the chromosome abnormality of a deleted 5q. Two other agents that are approved for intermediate-2–risk and high-risk MDS are hypomethylating agents azacitidine (Vidaza) and decitabine (Dacogen). The results of clinical trials have increased the understanding of these treatments alone or in combination with other therapies. Effective management often requires at least 3 to 6 months to achieve a clinical response; therefore supportive care is necessary.
Supportive care includes blood transfusions for anemia and platelet transfusions when platelet levels are very low. Erythropoietin stimulating proteins, such as epoetin alfa (Epogen, Procrit) or darbepoetin alfa (Aranesp) may be given in addition to transfusions. Frequent transfusions can lead to iron overload and organ damage.
Chelating therapy may be needed to reduce the iron overload before damage to the liver and heart occurs. Chelating is the chemical binding of iron and its removal from the body. It is indicated for patients who receive more than 20 units of red blood cell transfusions or for those whose serum ferritin levels are greater than 2500 mcg/L. Chelating drugs include deferasirox (Exjade) and deferoxamine mesylate (Desferal, Desferrioxamine).
White Blood Cell Disorders
As discussed in Chapter 19, white blood cells (WBCs), or leukocytes, provide protection from infection and cancer development. This protection depends on maintaining normal numbers and ratios of the different mature, circulating WBCs. When any one type of WBC is present in either abnormally high or abnormally low amounts, immune function is altered to some degree, as are oxygen transport and blood clotting, placing patients at risk for many complications. This section covers the changes and nursing care for patients with disorders involving overgrowth of specific types of WBCs. (See Chapter 21 for the problems and care needs for patients with immune deficiency.)
Leukemia
Pathophysiology
Leukemia is a type of cancer with uncontrolled production of immature WBCs (usually “blast” cells) in the bone marrow. As a result, the bone marrow becomes overcrowded with immature, nonfunctional cells and production of normal blood cells is greatly decreased. Leukemia may be acute, with a sudden onset and short duration, or chronic, with a slow onset and symptoms that persist for years.
Leukemias are classified by cell type. Leukemic cells coming from the lymphoid pathways (see Fig. 19-3 in Chapter 19) are typed as lymphocytic or lymphoblastic. Leukemias in which the abnormal cells come from the myeloid pathways are typed as myelocytic or myelogenous. Several subtypes exist for each of these diseases, which are classified according to the degree of maturity of the abnormal cell and the specific cell type involved. Biphenotypic leukemia is acute leukemia that shows both lymphocytic and myelocytic features.
With leukemia, cancer most often occurs in the stem cells or early precursor leukocyte cells, causing excessive growth of a specific type of immature leukocyte. In some chronic leukemias, the cancerous cells may be more mature. These cells are abnormal, and their excessive production in the bone marrow stops normal bone marrow production of RBCs, platelets, and mature leukocytes. Anemia, thrombocytopenia, and leukopenia result. Often the number of immature, abnormal WBCs (called “blasts”) in the blood is greatly elevated, and these cells cannot provide infection protection. Leukemic cells can also be found in the spleen, liver, lymph nodes, and central nervous system. Without treatment, the patient will die of infection or hemorrhage. For patients with acute leukemia, these changes occur rapidly and, without intervention, progress to death. Chronic leukemia may be present for years before changes appear.
Etiology and Genetic Risk
The exact cause of leukemia is unknown, although many genetic and environmental factors are involved in its development. The basic problem involves damage to genes controlling cell growth. This damage then changes cells from a normal to a malignant (cancer) state. Analysis of the bone marrow of a patient with acute leukemia shows abnormal chromosomes about 50% of the time (McCance et al., 2010). Possible risk factors for the development of leukemia include ionizing radiation, viral infection, exposure to chemicals and drugs, bone marrow hypoplasia (reduced production of blood cells), genetic factors, immunologic factors, environmental factors, and the interaction of these factors.
Ionizing radiation exposures such as radiation therapy for cancer treatment or heavy radiation exposure (e.g., the atomic bomb at Hiroshima or the nuclear accident at Chernobyl) increase the risk for leukemia development, particularly acute myelogenous leukemia (AML).
Chemicals and drugs have been linked to leukemia development because of their ability to damage DNA. Previous treatment for cancer with chemotherapy (e.g., melphalan, doxorubicin, etoposide, and cyclophosphamide) poses risks for leukemia development about 5 to 8 years after treatment. Table 41-3 in Chapter 41 lists chemicals and drugs that damage the hematologic system.
Bone marrow hypoplasia can increase leukemia risk by reducing or changing the rate of bone marrow cell production. Disorders that have marrow hypoplasia and may lead to leukemia development include Fanconi’s anemia and myelodysplastic syndromes.
Genetic factors influence leukemia development. There is an increased incidence of the disease among patients with hereditary conditions such as Down syndrome, Bloom syndrome, Klinefelter syndrome, and Fanconi’s anemia. Identical siblings of patients with leukemia have a higher incidence of leukemia than do the general population.
Immunologic factors, especially immune deficiencies, may promote the development of leukemia. Leukemia related to immune deficiency may be a result of loss of immune protection, or the same mechanisms that cause the immune deficiency may also trigger cancer in the WBCs.
Interaction of many host and environmental factors may result in leukemia. Because each person tolerates the interaction of these factors differently, it is difficult to determine the origin of any specific leukemia.
Incidence/Prevalence
Leukemia accounts for 2% of all new cases of cancer and 4% of all deaths from cancer (American Cancer Society, 2011). The incidence depends on many factors, including the type of WBC affected, age, gender, race, and geographic locale.
In the United States, about 44,000 new cases of leukemia occur each year (American Cancer Society, 2011). Leukemia is classified into any one of four types based on the cell type affected and how fast the disease progresses:
• Acute myelogenous leukemia (AML) is the most common form of adult-onset acute leukemia. There are eight subtypes of AML, which are diagnosed and classified on the basis of the number of healthy blood cells, the number of leukemic cells, and the specific chromosomal abnormalities identified in the leukemic cells. Disease prognosis varies by subtype and chromosomal abnormality. Acute promyelocytic leukemia (APL) is a common subtype of adult-onset AML that is now the most curable of the adult AMLs (Walker & Held-Warmkessel, 2010).
• Chronic myelogenous leukemia (CML) makes up about 20% of adult-onset leukemias, occurring more often in people older than 50 years. A major feature of CML is the presence of the Philadelphia chromosome abnormality in the leukemic cells. CML has three phases:
• Chronic lymphocytic leukemia (CLL) is the most common type of chronic leukemia and occurs most often in people older than 50 years (Elphee, 2008). Average survival time ranges from less than 19 months for patients diagnosed with advanced disease to more than 10 years for patients diagnosed with early-stage disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


