Following the fusion of the prominences the palates form to divide the primitive oral cavity into a separate nasal cavity and oral cavity. By day 51 the fusion is completed and the soft palate and uvula are formed by day 53.
General Care Principles
It is essential to have a basic working knowledge of the normal anatomy and physiology as well as the related developmental stages of the mouth as the child in PICU may have impaired functioning or related risks, for example oral intubation impairs the swallowing reflex and the presence and stimulation of an orally placed endotracheal tube can cause excess salivation. This will result in the children’s nurse having to make frequent assessment of salivary build-up and decide whether to start oral suctioning, mindful of the fact that this procedure can also cause stimulation which will increase secretions as well as disturb and frighten a lightly sedated child.
The procedure of intubation or the friction caused by the presence of an ETT can cause complications, such as the displacement of loose teeth. The presence of an endotracheal tube interrupts the normal routine of a child’s oral hygiene and although there is a paucity of evidence to support good practice, the management of a child in PIC must include consideration of oral hygiene to prevent infection or the risks associated with descending infection resulting in ventilator associated pneumonia (Cutler and Davis 2005; Pobo et al. 2009; Somal and Darby 2006; Wikin 2002) (Chapter 4).
Pre-Operative Fasting
The RCN guidelines on pre-operative fasting (2005) focus on healthy infants and children and although there is some comparison where major elective surgery is concerned (e.g.an infant or child who is fit and well and going to be recovered following surgery on PICU), most pre-operative fasting guidelines now permit the intake of water and other clear fluids up to 2 hours before the induction of anaesthesia for elective surgery in healthy infants and children in order to prevent the risks of excessive pre-operative fasting and enhance the child’s wellbeing. The children’s nurse needs to be aware that some surgical procedures are lengthy and children may have been fasted for prolonged periods by the time they reach PICU. The same guidelines do not apply to children who have been traumatised and are in a shocked state. In addition, where emergency admissions are concerned, the history of when the infant was last fed or the child last ate may be vague. The decision to proceed with urgent surgery needs to be made in the child’s best interests.
There is a high correlation between malnutrition and outcome, yet fasting and feeding practices for children in PIC are not based on good evidence. As there is more evidence available to support the neonatal nurse this can be used to support infant gut management. There is also good quality work informing general intensive care practice and this can be used to support the management of the adolescent and the young person. Where possible appropriate evidence has been cited but there is a distinct gap in the evidence base covering the intermediate age range. Intubation and ventilation can delay the gastric emptying time and this has been associated with increased gastric residual volumes. It is known that morphine administration affects antroduodenal motility as the gastrointestinal motor pattern involved is characterised by antral hypomotility (Bosscha et al. 1998); however, there is little information on normal gastric residual volume in paediatric patients (Babbitt 2007). An awareness of gastric emptying in critically ill children is important as the consequences of this phenomenon have implications for intolerance to enteral nutrition, the range and extent of gastric colonisation and an increased risk of aspiration pneumonia (Heyland et al. 1996; Moreira and McQuiggan 2009). (See Chapter 12 for complex fluid and feeding management.)
Stress Ulceration
Stress ulcers of the stomach and duodenum, as well as upper gastrointestinal (UGI) bleeding are well-known complications of critical illness in children who are admitted to a PICU. Calculations of the prevalence of stress ulceration are variable, with the incidence increasing with the duration of stay and there being a correlation with the development of ulceration and risk of mortality. Prophylaxis against stress ulcers is the preferred form of management, using a range of strategies, pharmaceutical agents and a variety of dosage and regimens:
- Early feeding.
- Omeprazole.
- Ranitidine.
- Sucralfate.
- Famotidine.
- Amalgate.
- Antacids.
Further work requires to be done to develop good guidance and treatment protocols (Reveiz et al. 2010).
Medical Management of Acute or Life-Threatening Abdominal Conditions
Pancreatitis
Pancreatitis, although uncommon during childhood, is associated with significant morbidity and mortality. This condition is characterised by inflammation of the pancreas, clinical signs of epigastric abdominal pain and elevated serum digestive enzymes. Pancreatitis can be local or diffuse and is classified as acute, chronic, inherited, necrotic or haemorrhagic. Occasionally, pancreatitis is complicated by the formation of a fibrous-walled cavity filled with pancreatic enzymes, termed a pseudocyst (Chang et al. 2011).
Pancreatitis occurs as a consequence of the blockage or disruption of the collecting ducts and damage to the pancreatic acinar cells, which leads to activation and then the release of digestive enzymes. The activated enzymes autodigest the pancreatic parenchyma, causing inflammation and necrosis.
Pancreatitis may follow blunt abdominal trauma, occur as a result of congenital abnormalities in the pancreatic ducts, as a complication following a ‘mild’ viral ailment or as a serious side-effect of a powerful drug such as asparaginase, a chemotherapy drug. Asparaginase is an enzyme that breaks down protein, but fortunately this is a rare if serious side-effect. Children with cystic fibrosis may develop a chronic pancreatitis. Hereditary pancreatitis is an autosomal dominant disorder with an 80% penetrance, accounting for about 1% of cases (Obideen et al. 2008).
Acute haemorrhagic pancreatitis rarely occurs in children. This is a life-threatening condition with a mortality rate approaching 50% because of shock, systemic inflammatory response syndrome with multiple-organ dysfunction, acute respiratory distress syndrome (ARDS), disseminated intravascular coagulation (DIC), massive gastrointestinal bleeding and systemic or peritoneal infection (Werlin 2003).
Physical examination findings associated with haemorrhagic pancreatitis may include a bluish discoloration of the flanks (Grey Turner sign) or peri-umbilical region (Cullen sign) because of blood accumulation in the fascial planes of the abdomen. Additional signs may include the accumulations of pleural effusions, haematemesis, melaena and coma (Chang et al. 2011; Werlin 2003).
Clinical Presentation of Acute Pancreatitis
- Severe pain located in the epigastric or para-umbilical regions, with the child adopting a foetal position with their knees drawn up to try to relieve this.
- Abdominal distension.
- Presence of abnormal bowel sounds.
- Nausea and vomiting.
- Unexplained fever.
- Unstable serum blood glucose values.
- Elevated pancreatic enzymes.
Treatment
The goal of medical management of acute pancreatitis is to achieve adequate rehydration, analgesia and pancreatic rest and to restore normal metabolic homeostasis. In patients with severe pancreatitis, oral intake is usually restricted and total parenteral nutrition should be initiated within 3 days to prevent catabolism. There is considerable discussion in the literature about the point at which enteral nutrition should be reinstituted, but currently there is no consensus. In cases of intractable vomiting or ileus, a nasogastric tube left to free drain is helpful in promoting intestinal–pancreatic rest by eliminating gastric secretions in the duodenum, the most potent activator of pancreatic secretion. Fluid, electrolyte and mineral imbalances should be corrected urgently. Antibiotic therapy is indicated for systemic infections or sepsis. Acute pancreatitis should resolve in 2–7 days with adequate resuscitation (Benifla and Weizman 2003; Chang et al. 2011).
In chronic relapsing pancreatitis, pancreatic enzyme supplementation, insulin and elemental or low-fat diets are useful adjuncts to maximise the child’s nutritional status. For the alleviation of pain, opioids are essentially contraindicated as they can contribute to spasms of the sphincter of Oddi. Advice should be gained from a specialist pain team.
Crohn’s Disease
Crohn’s disease (CD) is an inflammatory bowel disease. It is a chronic condition characterised by repeated exacerbations and periods of remission. It usually involves the small intestine, most often the lower part (the ileum). However, inflammation may also affect the entire digestive tract, including the mouth, oesophagus, stomach, duodenum, appendix or anus. The cause is unknown, however there are several theories. One suggests that a trigger, possibly a microorganism, affects the body’s immune system and this results in an inflammatory reaction in the intestinal wall. There is supporting evidence that children with the disease have abnormalities of the immune system, however it is uncertain whether the immune problems are a cause or a result of the disease.
Pharmacological Agents Used in the Management of Crohn’s Disease
Drug therapy singly or in combination is the first-line approach to management. 5-aminosalicylates (5-ASAs) are a group of compounds that have long-established use in inflammatory bowel disease. However, while sulphasalazine may have a role in the treatment of mild CD, the overall evidence does not support the use of 5-ASA agents for inducing remission in CD (Akobeng 2008).
Antibiotics
Although there is no convincing and reproducible evidence, a number of antibiotics have been tried for the induction of remission in CD. However, the most rigorous available evidence does not support the routine use of antibiotics for the induction of remission in active CD (Akobeng 2008). Immunosuppressive agents such as azathioprine and 6-mercaptpopurine are purine analogues that have demonstrated effectiveness for the treatment of CD and in establishing remission.
Methotrexate
No controlled trials have been performed in children, but a retrospective case note analysis has suggested that methotrexate may be effective and safe for inducing remission in paediatric Crohn’s disease (Weiss et al. 2009).
Corticosteroids
Corticosteroids are also effective for inducing remission but may be associated with significant adverse events. Infliximab is recommended for the treatment of patients with severe Crohn’s disease who do not respond to conventional management (NICE 2002).
Symptomatic control may be achieved with the management of diet and analgesic regimes. There is interest in probiotic use, although the supportive action of probiotics is as yet not fully understood (Patel and Lin 2010).
Extracorporeal Photopheresis for Inflammatory Bowel Disease
Under research and awaiting clinical trials is the procedure called extracorporeal photopheresis. This is when the patient is anticoagulated with heparin and blood is removed from the patient using a two-way circulatory device to provide access to the circulation and allows the circulation to be returned. Once removed the blood is filtered and the white blood cells are separated from the whole blood. These white cells are treated with ultraviolet light and are then returned to the patient where they produce a generalised immune response against the pathogenic T-cell clones that are involved in the pathogenesis of inflammation in Crohn’s disease (NICE 2009). Extracorporeal photopheresis is usually carried out over two consecutive days at intervals of 2–4 weeks for about 20 treatment sessions. One ECP session takes about 3–4 hours. The therapy has a number of side-effects and it should only be performed as a last resort and as part of a research programme.
It is rare for these children to be admitted to a PICU unless in a life-threatening condition or to facilitate close postoperative monitoring and management.
Commonalities across Gastrointestinal Conditions, Surgery or Major Gut Insult
Peritonitis
This is inflammation or infection of the peritoneum; the inflammation can be caused by a blood-borne bacterial or fungal infection or be a result of perforation of the gut or digestive organs. It can be seen as a complication in children who are on chronic peritoneal dialysis (Chapter 6), or as a result of blunt trauma causing tears in fragile abdominal organs resulting in the spillage of gut contents into the abdominal cavity. However, it can also be caused by other conditions that allow bacteria, enzymes or bile into the peritoneum from damage to the gastrointestinal or biliary tracts. Examples include pancreatitis, a perforated appendix (the most common), stomach ulcer, Crohn’s disease or diverticulitis.
Signs of Peritonitis
- Swelling and tenderness in the abdomen with pain ranging from dull aches to severe, sharp pain.
- Pyrexia.
- Anorexia.
- Nausea and vomiting.
- Limited urine output.
- Ileus – silent abdomen, no flatus or stool.
Management of Peritonitis
Treatment and management is surgical repair where this was the primary cause and broad spectrum antibiotics. The gut needs to be rested and enteral fluids and feeds re-graded with care. The child needs effective management of pain. Drains are not routinely used in the management of post-surgical intervention in children but these children are at a relatively high risk of intra-abdominal abscesses (van Wijck et al. 2010), so rather than repeated trips to theatre some external drainage of the abscess bed may be considered.
Paralytic Ileus
This is a disruption of normal intestinal peristalsis, the natural gastrointestinal motor activity. Paralytic ileus is common following surgery where the gut is handled or the abdominal cavity has been opened, for example, to access the retroperitoneal space for renal surgery. It can also result from certain drugs such as anaesthetics and from major injuries where there has been circulatory impairment and poor perfusion. In children it can also be caused by sepsis. Paralytic ileus causes total constipation, absence of flatus and abdominal distension. Auscultation with a stethoscope will reveal a silent abdomen as there are no borborygmi since the bowel is inactive. Despite treatment and management a small proportion of children go on to develop sub-acute pseudo-obstruction syndromes (Barr 1998).
‘Drip and Suck’
There is controversy about the requirement for gastric and gut decompression with a nasogastric tube (NGT) in abdominal surgery patients, in many cases it seems to be performed routinely in spite of the absence of good evidence (Moreira and McQuiggan 2009; Williams and Leslie 2004). Dinsmore and colleagues (2004) suggest that it is an unnecessary intervention in most cases. However, if paralytic ileus is present and gastrointestinal secretions are pooling in the stomach and gut loops, aspiration of these is an appropriate nursing action, particularly if the build-up of gastrointestinal secretions could cause vomiting and risk aspiration. In addition, aspiration of these secretions may avoid leakage or spillage through a perforation and their removal may reduce intestinal intra-lumen tension and help protect a new anastomosis. Sometimes the NGT is left on free drainage/siphon drainage. The volumes and character of all aspirated fluids need to be charted and as this is fluid lost to the child, a clear strategy of replacement fluid and electrolytes implemented.
The strategies for managing paralytic ileus are to rest the gut and allow it to recover, to prevent further complications and to administer sufficient fluids and calories to allow repair, recovery growth and development. In some cases this will mean long-term parenteral nutrition (Chapter 12).
Managing gut motility problems, promoting absorption and preventing long-term sequelae need to be done with the full involvement of the paediatric dietetic team and where indicated, the speech and language department. In self-ventilating small infants or those supported via a tracheostomy there is a need for oral stimulation and non-nutritive sucking if a good long-term outcome is to be achieved, as these infants may not have established feeding by mouth and, where the mouth has been used to access the airway during intubation procedures, steps need to be taken to avoid oral aversion, etc. Although controversial, a strategy of ‘kick-starting’ the gut may be tried in order to stimulate it into motility and acclimatise it to holding volume and absorbing elements. Both Miedema and Johnson (2003) and Stewart and Waxman (2007) reviewed additional supportive methods for stimulating gut activity and although there is a requirement for paediatric evidence to be built up, ultimately the adoption of prokinetic pharmaceutical support to enhance motility may shorten the duration of the ileus (Deane et al. 2009).
Necrotising Enterocolitis (NEC)
This is primarily a disease of the premature infant and is the most common gastrointestinal emergency in neonates. The aetiology is uncertain; however the smaller and sicker the infant, the greater the risk. Suggested contributing factors include the immature gastrointestinal anatomy and physiology. An immature intestinal barrier may permit bacteria to penetrate the mucosal barrier and trigger an inflammatory response. Immature biochemical defences may also be a factor where bacterial overgrowth is permitted and this may attack the bowel wall. Other contributory factors include hypoxic-ischaemic injury, early feeding with formula milk, not having breast milk and colonisation by pathological bacteria (Schnabl et al. 2008).
The epidemiology of NEC is little understood, with cases seemingly coming in clusters and some infants succumb who would seem not to have the predisposing factors of immaturity or a compromised gut because of a difficult postnatal phase, with serious RDS and early artificial feeding. Gut ischaemia and ileus in older infants and children can result in a toxic state where prolonged ileus can allow enteric bacteria to ferment malabsorbed carbohydrates to various gases, producing distension and increase the intraluminal pressure. This can result in a vicious cycle where distension of gut loops can further decrease the mucosal blood flow. The products of fermentation could be toxic to enterocytes and impair the mucosal barrier function. The inflammatory response can be triggered and the gut walls become very friable. There may be a risk of perforation and life-threatening peritonitis can set in.
Neonates with cyanotic heart disease also have a higher risk of developing NEC. While the pathophysiology of this is not completely determined, it is thought to be related to hypoxic injury (secondary to cyanotic heart lesion) to the immature GIT. NEC is a serious complication and can be staged based on clinical presentation and radiological findings (Table 8.3).
Table 8.3 Stages of necrotising enterocolitis
Source: modified from Bell et al. 1978; Walsh and Kliegman 1986.
| Stage | Impact on the infant |
| Stage 1 Suspected NEC | Systemic but non-specific signs such as thermal instability, lethargy, apnoea, bradycardia, not being themselves. Feeding intolerance with increasing gastric aspirates and blood in the stool. Abdominal X-ray may be normal. Management: medical as described below and await progression or resolution. |
| Stage 2(a) Mild NEC | Systemic non-specific signs; unstable, increasing apnoea and bradycardia which may require PICU management. Vomiting, abdominal distension, pain on examination, silent abdomen, frank blood in stool. Abdominal X-ray confirms ileus and local pneumatosis intestinalis. Management: continue medical management and refer for surgical opinion. |
| Stage 2(b) Moderate NEC | Systemically increasingly unwell with mild acidosis, thrombocytopenia. Abdominal wall oedema and tenderness with or without mass. Abdominal X-ray shows extensive pneumatosis intestinalis, early ascites, with or without intrahepatic portal gas. |
| Stage 3(a) Advanced NEC | Systemically very unwell with a respiratory/metabolic acidosis, apnoea, hypotension, decreasing urine output, leucopenia and disseminated intravascular coagulation (DIC). GIT: spreading oedema, erythema, induration of the abdomen. Abdominal X-ray shows prominent ascites with or without persistent sentinel loop but no perforation. Consent for surgery and monitor very closely. Transfusion and blood products. |
| Stage 3 (b) Advanced NEC | Systemically demonstrating clinical deterioration, shock, electrolyte imbalance. Abdominal X-ray shows signs of perforation. Requires urgent surgery. |
NEC – Strategies for Reducing the Risk
Neonatologists have developed a strategy where small amount of ‘gut priming’ or trophic feeds are given in order to avert adverse events, although Bombell and McGuire (2009) conclude that there was insufficient evidence to justify the practice of withholding or initiating early feeds. Gut stimulation may also prevent fasting-induced mucosal atrophy, so preventing bacterial translocation and the risk of endotoxaemia, mucosal inflammation or sepsis (Premji and Chessell 2007). It is important to remember that these ‘feeds’ are not given for nutritional value but to act as a protective function, and by giving the gut secretions something else to work on, may help prevent mucosal damage. Patole and de Klerk (2005) note a significant decline in the incidence of NEC following the implementation of a standardised feeding regime.
Reducing the risk of NEC has resulted in studies which employ probiotics and there is some evidence of their effectiveness (Alfaleh et al. 2010). Although there is emerging support for the use of oral lactoferrin, which is a normal component of human colostrum, milk, tears and saliva in enhancing the infant’s host defence in the prevention of sepsis, there is, as yet, no evidence that it reduces the incidence of NEC (Pammi and Abrams 2011).
Preventing mucosal damage is also part of the controversial theory behind the prophylactic prescribing of a histamine receptor blocker, for example IV ranitidine or cimetidine. These drugs work by blocking H2 receptors found on the cells in the stomach lining. Histamine normally binds to these receptors, causing the cells to produce stomach acid. Blocking the H2 receptors, prevents histamine from binding to them. This stops the cells from producing stomach acid which erodes the mucosa, producing ulceration and occasionally causing major haemorrhage. However, all drugs have side-effects. Consequently, Messori and colleagues (2000) recommend the use of H2 blockers on the basis of symptoms only, as there may be a link with their routine use and atypical pneumonia; and Donowitz and colleagues(1986) note the abnormal gastric colonisation of patients with therapeutically altered gastric acidity by hospital- acquired gram-negative rods.
Medical Management of NEC
There is an element of repetition in the strategy for managing NEC medically in common with other intestinal failures:
- Enteral feeding is stopped so the gut is rested.
- NGT placement – this is usually left on free drainage with regular aspiration for gastrointestinal decompression and the infant’s comfort.
- Excessive gastric losses may need to be replaced (millilitre for millilitre) with 0.9% sodium chloride +/− potassium supplementation to avoid the infant becoming hypokalaemic or hyponatraemic.
- PN and complex fluids are titrated to the individual infant’s needs (Chapter 12).
- Triple antibiotic therapy, including metronidazole.
- Analgesics.
- Supportive management.
- Minimal handling.
Surgical Management of NEC
There is a general reluctance to expose fragile infants to the trauma of surgery; however some who are not recovered by medical means are so sick that surgery becomes their only option for survival (Pierro and Hall 2003). Tepas and colleagues (2010) consider some metrics of metabolic derangement which could be used as indicators to support the clinical decision to intervene surgically.
There are two main surgical options: laparotomy, visual inspection, resection and anastomosis; and the less invasive paracentesis and primary peritoneal drainage. Moss and colleagues (2006) consider one option as having a greater advantage than the other. Other units have used a combination of methods, with the drainage sometimes being used as an interim method to try to stabilise the infant before laparotomy. However Rees and colleagues (2010) did not find that peritoneal drainage improved clinical stability in infants where there was bowel perforation.
If the infant requires a laparotomy, it is difficult to prepare the parents as to how the infant may look following surgery and whether they will have an ileostomy or not. If a single area of bowel is affected and resected and the remainder of the bowel is in good condition, a primary anastomosis may be possible. However, this exposes the infant to the risk of leakage, stricture, fistula or breakdown. More frequently a proximal ostomy and distal mucous fistula are created, which may be reversed once the infant’s condition improves.
Vaughan et al. (1996) refined a technique of clipping and closing where necrotic segments of intestine were resected and the transected ends stapled closed. This meant that the infant had to return to theatre 2–3 days later and have a reanastomosis. If all went well, however, these infants did not have a stoma.
Surgeons sometimes have to make some very difficult choices. When less than 25% of the intestinal length is found to be unaffected the choices are restricted. Simple closure results in a very poor outcome and most surgeons opt for patching where they can, drain, hope and wait.
The prognosis is variable in infants with surgically managed NEC and can differ according to the location of the NEC. The infants who had resection for NEC in the large bowel did better than infants who had resection in the short bowel (Zhang et al. 2011). This also had implications for mortality, co-morbidities, length of stay and the cost of the hospital stay.
Short Gut Syndrome and Short Bowel Syndrome (SBS)
SBS can be defined as a malabsorptive state resulting from a congenital malformation of the gut or occurring after an extensive resection of the small intestine (Sala et al. 2010) or for acquired lesions such as intestinal volvulus resulting in ischaemia (Duro et al. 2008). Regardless of the primary cause, the gut usually compensates by dilating to create more surface area to absorb nutrients. This slows down food transit, can increase the amount of bacteria in situ and can result in potentially life-threatening infections. Where children cannot absorb their nutrients, Parenteral nutrition (PN) will be employed to sustain growth. However, this places the child at risk of PN-associated complications resulting in more pathology.
Strategies to Manage Short Bowel Syndrome
Wherever possible the remaining gut should be put to use and refeeding introduced:
- Parenteral nutrition with vitamin and mineral replacement.
- Careful fluid management.
- Monitor for and manage gastric acid hypersecretion.
- Management of the complexities of malabsorption.
- Management of motility.
- Management of any bacterial overgrowth with appropriate antibiotics.
- Surgical lengthening procedures and transplant.
Some of these children are frequently exposed to a range of medications in the hope of improving the residual gut function. There is a paucity of evidence regarding the application of pro-motility and anti-diarrhoeal medications in patients with SBS and intestinal failure, although they continue to be used (Dicken et al. 2011).
Refeeding Collected Gut Secretions
High stomas located in the jejunum or proximal ileum can result in the production of large quantities of effluent containing unabsorbed nutrients resulting in poor growth and electrolyte imbalance. The neonate is usually dependent on PN until the intestinal adaptation takes place; however PN is associated with significant risk of central line-related sepsis, thrombosis and neonatal cholestasis. Collecting the effluent from the high stoma and refeeding this effluent into the distal mucous fistula uses the absorptive surface of the distal bowel for nutrient absorption, may stimulate mucosal growth, improve intestinal adaptation and prevent atrophy of the distal bowel. This should result in weight gain, reduce the electrolyte imbalance and decrease dependency on PN (Richardson et al. 2006). The procedure is simple to perform and may be done by parents following discharge. However, the effluent has an unappealing character and this strategy needs considerable explanation to the parents to allow them to come to terms with it.
Surgery
SBS has been treated with a surgical process called the Bianchi procedure. During this, the bowel is bisected and one end is sewn to the other. However, the bowel often redilates, leaving patients in the same condition as when they started. Serial transverse enteroplasty has the potential to avoid these difficulties and the procedure involves stapling V shapes into alternating sides of the bowel resulting in a decrease of width and an increase in length (NICE 2007). Research supports an improvement in bowel function and nutrition (Kajia et al. 2009) and it is possible the procedure will become more commonplace and lend itself to other applications (Modi et al. 2007).
Intestinal Transplant
This is now a well-recognised alternative treatment strategy for SBS (Grant et al. 2005). Although most will come from cadaveric donation there is an interest and an increase in living related donations (Li et al. 2008). Graft choice depends on the presence of associated liver disease (Duro et al. 2008) as a block graft including liver may be preferred to isolated bowel graft. Although puberty can be delayed in some cases, a functional intestinal graft can result in a normal growth pattern; the exception seems to be in children who remain on high-dose steroids (Lacaille et al. 2007). Post-transplant children require life-long anti-rejection management as the gut is an immune organ harbouring a large amount of immune-competent cells, has a large colony of microorganisms (Braun et al. 2007) and it is particularly vulnerable to the effects of ischaemic injury. This susceptibility to damage will not permit time-consuming HLA matching prior to transplantation (Ruemmele et al. 2006). Although protocols differ (Pirenne and Kawai 2006) anti-rejection management may include starting therapy before the transplant to kill or deplete T- and B-cells which would target the transplanted intestine.
Following transplantation, the immunosuppressive therapy such as tacrolimus and tapered corticosteroid doses (Martingale 2009) continues, although there seems to be controversy regarding the use of steroids (Dazzi et al. 2007). Chronic rejection does not appear to develop insidiously in compliant patients and although rare, late acute rejection may complicate infectious diarrhoea (Lacaille et al. 2007).
Stoma Formation and Stoma Care
A stoma simply means an opening. Paediatric ostomies include any surgically created opening between a hollow organ and the skin connected either directly (stoma) or in the case of gastrostomy with the use of a tube or peg. The following are examples of stoma and are not an exhaustive list:
- Gastrostomy usually created for enteral feeding.
- Duodenostomy usually created for enteral feeding.
- Jejunostomy.
- Ileostomy.
- Proximal colostomies.
In infants and children, stomas are used for various purposes, including access, decompression, diversion and evacuation (Minkes et al. 2008). Most ostomies in children are for temporary use and are typically reversible after a determined period of time, although some medical conditions may dictate the need for a permanent stoma (Minkes et al. 2008). Complications following intestinal diversional (ostomy) surgeries are high (Park et al. 1999) and can present a significant problem to many individuals. It has been estimated that up to 70% of patients with an ileostomy and 43% of those with a colostomy experience complications (Pittman and Rawl 2009).
There are several differences between adult and paediatric ostomies. In adults most stomas are formed in the distal ileum or colon for the treatment of inflammatory bowel disease, malignant conditions or trauma. In infants and children a stoma may be required anywhere along the GI tract because of a range and variety of congenital and acquired conditions. The effects of stoma formation for the infant or child should not be underestimated. They include the impact on the parents, which can affect bonding in the neonatal period, the impact on the family of repeated hospitalisations, which can affect other siblings, and the effects that a stoma may have on the child’s physical and emotional development. The physiological impact may affect a child’s growth and ability to maintain their own nutritional needs, while the emotional and psychological effects, particularly in the older child or adolescent, require careful management. There is potential for social isolation, with some children being reluctant to attend school or engage with their peer group; referral to a specialist nurse is recommended.
Many stomas in infants and children are undertaken as emergency procedures, however whenever possible a primary stoma site and back-up sites should be selected and marked before surgery. The stoma should be distant from the incision and away from skinfolds, bony prominences and umbilicus. Treating a child with multiple abdominal stomas can be challenging, especially when the anatomy is unclear and the fluid and electrolyte abnormalities are difficult to manage (Minkes et al. 2008).
Small bowel stomas, such as a jejunostomy, ileostomy or a proximal colostomy, have very liquid output and the volumes of effluent may be large. The stoma output can contain enzymes and other bowel contents which have the potential to irritate the skin. Skin irritation, excoriation and infection are among the most common complications with paediatric stomas. To help prevent this, the use of an appropriately fitting stoma appliance is essential. Various styles of appliance are available and there is an increasing range of barrier creams and products to select from.
Major Surgery
Indications for Major Surgery
It is estimated that about 75% of individuals who live with Crohn’s disease will require surgery at some point and that 75% of those who have one surgery will need at least one subsequent surgery. Many of these procedures will be minor, however some can be life-threatening. In life-threatening inflammatory bowel disorder, surgery is indicated:
- Where the bowel has become irreparably damaged.
- Where there is necrotic tissue causing toxaemia.
- Where a stricture has formed resulting in obstruction.
- To drain large and deep abscesses.
- To correct defects where there are multiple fistula.
Surgical approaches include conventional laparotomy, minimally invasive surgical techniques or endoscopic procedures. Increasingly, major procedures will be performed using a minimally invasive surgical procedure or an endoscopic surgical approach. However, at present only a few paediatric procedures are routinely offered, or can be performed using the minimally invasive approach. The laparotomy technique is used to expose the majority of the abdominal organs which allows for confirmation or correction of the preoperative diagnosis in a child presenting with an acute abdomen.
Minimally Invasive Surgery
Jaffray (2005) states that minimal access techniques are sought to perform a range of surgical procedures while avoiding the morbidity of conventional surgical wounds. This was first developed in the mid-1980s and became possible following three technological advances: a system allowing instrumental access into the body cavities; the scaling down of video cameras which provided good visualisation inside the body; and the production of insufflation devices to allow controlled distension of body cavities with gas to provide the surgeon opportunity to see and space to work.
The advantages of minimally invasive techniques include avoiding large wounds and the need to cut through abdominal muscles which results in less pain and the complications of immobility. Less pain facilitates earlier discharge and shorter absences from school. There is also a better cosmetic result and less risk of infection.
Endoscopic Approach
This is one of the fastest-growing fields of surgery. The flexible endoscope containing some form of illumination (usually a fibre-optic light source) which also contains a small channel for instruments to enable procedures such as taking biopsies and the retrieval of foreign bodies is appealing and effective for minor paediatric procedures. However, its use is limited owing to the small size of the gut lumen in the infant population.
Further developments in paediatric surgery and the flexibility of endoscopic equipment should allow for expansion of both these approaches in children. Trials underway with animal studies are being carried out to assess the possibilities of a natural orifice endoscopic minimally invasive approach (ASERNIP-S Report 2007).
Congenital Corrective Surgery
While the majority of infants requiring congenital corrective surgery will be managed in the neonatal intensive care unit, a number of usually full-term infants with potentially life-threatening defects will be managed in the paediatric intensive care setting as a consequence of cot shortages in the neonatal unit. On occasions, units may also need to accommodate pre-term infants and managing these infants can be a challenge as they have different requirements in terms of fluid management strategies, thermoregulation and pain control. In addition, the parents are in a uniquely vulnerable situation as the mother may be unwell herself following delivery and the father may have divided loyalties, having to allocate his time between the sick infant, his partner and any other children.
Tracheoesophageal Fistula and Oesophageal Atresia
Tracheoesophageal fistula is very commonly associated with oesophageal atresia (OA) and therefore the lesions will be discussed together. The abbreviation TOF referring to this condition should not be confused with tetralogy of Fallot which is also referred to as TOF in some sources. TEF is the abbreviation used in the United States for this condition and may minimise confusion.
A tracheoesophageal fistula is a congenital (or occasionally acquired) communication between the trachea and oesophagus, while oesophageal atresia refers to a congenitally interrupted oesophagus. A tracheoesophageal fistula/OA will occur as a consequence of the failure of the mesenchymal separation of the upper foregut, but there is no fixed time for this to happen. It affects more males than females and between 10 and 40% of infants in the reported series are preterm. Between 35 and 65% of these infants will have associated anomalies such as congenital heart disease, VATER syndrome or VACTERL association (Diaz et al. 2005).
There are five commonly identified variants of tracheoesophageal fistula +/− OA which are detailed in Table 8.4.
Table 8.4 Variants of tracheoesophageal fistula and OA presentation
| Type | Characteristics of lesion |
| A | OA with distal tracheoesophageal fistula (most common variant) |
| B | Pure OA with no tracheal involvement |
| C | Isolated tracheoesophageal fistula with no oesophageal involvement |
| D | OA with proximal tracheoesophageal fistula |
| E | OA with both distal and proximal tracheoesophageal fistula (least common variant) |
Clinical Presentation of TOF/OA
- Choking on first feed.
- Coughing.
- Cyanosis.
- Excessive salivation.
- Aspiration pneumonia.
Chest X-ray Findings
- Coiled orogastric tube in the cervical pouch; air in the stomach and intestine.
Pre-Operative Management
The major aim of preoperative management is to minimise or prevent any pulmonary complications which may occur as a consequence of the aspiration of gastric or oesophageal pouch contents. The infant is kept nil by mouth and nursed in a cot or babytherm on a tilt to ensure that a head-up position is maintained. A Repogle tube (a double lumen, radio-opaque tube) is sited into the blind-end oesophageal pouch, left on low continuous suction and flushed regularly with a small volume (0.5 ml) of 0.9% sodium chloride to maintain patency. Guidelines for the frequency of flushing should be available from the neonatal unit with intervals varying between 15 and 30 minutes. The thickness of secretions will determine how regularly the Repogle tube requires flushing.
Surgical Management
The type of surgical approach will be determined by the size of the gap between the two ends of the oesophagus (Table 8.5). A primary repair is possible when there is a small gap and reanastomosis will not cause undue tension on the site of repair when the infant moves. Delayed primary repair may be required if there is a long gap OA (<the distance between six vertebrae) or if the gap is greater than six vertebrae then oesophageal substitution may be necessary.
Table 8.5 Management approaches for TOF/OA
| Primary repair | Delayed primary repair | Oesophageal substitution |
| Intubation/ventilation for up to 5 days Head midline and use of muscle relaxants to avoid tension on the suture line Feed via a trans-anastomotic tube (TAT) Barium swallow prior to commencing oral feeds (to check for anastomotic leak) | Gastrostomy Long-term Repogle tube placement/management Re-imaging to determine growth Use of cervical oesophagostomy and sham feeding will allow infant to develop feeding skills during this time Adequate growth – postoperative care as per primary repair | Management as per delayed primary repair and then surgical approach of:
|
Colonic Interposition
A section of colon is transposed, with its blood supply intact, into the chest where it is joined to the oesophagus and the stomach bridging the gap. The advantages of this procedure are that the size of the graft is of equal diameter to the missing oesophagus and there is no limit to the size of the graft required (Thomas et al. 2009). The associated complications, however, need to be considered carefully:
- The blood supply to the transplanted section of colon is precarious.
- Poor peristalsis can lead to feeding difficulties.
- High incidence of leakage (30%).
- Stricture (narrowing) can occur (20%).
- Redundancy (lack of any muscular activity) can develop in the long term.
Gastric Tube Oesophagoplasty
A longitudinal segment is taken from the stomach, which is then positioned into the chest and joined to the oesophagus. The advantages include a good blood supply to the graft and the size of the graft is appropriate to the infant or child. Complications again need careful consideration as there is a very long suture line which leads to an increased risk of leakage and a high stricture rate. In addition, there is a significant risk of reflux disease.
Gastric Transposition
This is a relatively new procedure in which the whole stomach is freed, mobilised and moved into the chest. The upper end of the oesophagus is then anastomosed to the top of the stomach in the neck (Hirschl et al. 2002). Advantages of this procedure are the lack of a long suture line, which minimises the risk of both leakage and stricture formation. The long-term consequences are not yet clearly identified, however, short-term complications include:
- Poor gastric emptying.
- As the bulk of the stomach is in the chest, respiratory capacity is reduced.
- Reflux can be a problem.
- ‘Dumping syndrome’ occurs when food enters the intestine relatively quickly and causes sweating, dizziness and diarrhoea and blood glucose imbalances.
Long-Term Consequences of TOF/OA
- Persistent cough.
- Gastro-oesophageal reflux disease (GORD).
- Recurrent lower respiratory tract infections.
- Strictures.
- Feeding difficulties.
Duodenal Atresia
This is the most common foetal atresia, occurring in 1/5 000 births and is associated with Down’s syndrome. It is a condition in which a part of the GIT tract from the duodenum to the anus has failed to form correctly and that part of the gut is either completely blocked or is missing altogether. This can result in life-threatening obstruction and the defect can only be corrected surgically.
There are three main types:
- Type I: Obstruction due to a mucosal web with normal muscular wall.
- Type II: Two atretic duodenal ends joined by a short fibrous cord.
- Type III: Complete separation of atretic ends with no connective tissue.
Many cases are now picked up on antenatal scans as a ‘double bubble’ may appear on scan due to the dilated, fluid-filled stomach and proximal duodenum. Parental counselling, advice and support should be offered at this point to ensure that they are adequately prepared. The pregnancy may be complicated by polyhydramnios owing to the impaired absorption of amniotic fluid by the foetal intestines. The defect may be a single one or complicated by intestinal malrotation and congenital heart disease. Postnatally, it may be suspected by the presence of vomiting within hours of birth. This vomit is most often bilious, although it may be non-bilious because a small number of atresia occurs proximal to the ampulla of Vater. Because the gut is not patent the infant might have a ‘hollow’-looking abdomen (scaphoid). An X-ray may demonstrate a gas-filled ‘double bubble’ which corresponds to the antenatal fluid-filled image and, unless there is perforation, there is no other gas present in the intestine or abdominal cavity.
Pre-Operative Management
Although the condition is potentially life-threatening it is not an emergency and, if otherwise well, the infant can be left for between 24 and 48 hours before undergoing surgery. The infant must remain nil by mouth and be maintained with IV hydration. An NGT should be passed and the gut aspirated. If prolonged NGT aspiration is necessary or the amounts copious, the IV regime should include replacement of the gastric aspirate with either 0.45% or 0.9% sodium chloride. Prior to surgical repair, the infant’s fluid and electrolyte status must be checked.
Surgical Management
This has been performed laparoscopically and the techniques are being continually refined (Valusek et al. 2007). However, a laparotomy and a duodenostomy are the most commonly performed procedures. There are a couple of techniques for this and they are used at the surgeon’s preference, but essentially they involve opening the duodenum channel along its length and joining it to the next portion of patent intestine, and correcting the duodenal lumen end to end so that a fully open channel exists. Most surgeons place a small trans-anastomotic (TAT) feeding tube to protect the suture line and expedite gut priming; some evidence suggests that toleration of enteral feeds is quicker as a result (Arnbjörnsson et al. 2002). The entire small bowel is carefully explored for other sites of obstruction, the hepatic and pancreatic anatomy is checked and any malrotation corrected.
Postoperative Management
During the immediate postoperative phase the infant is usually intubated and ventilated. This maintains the airway and provides adequate oxygenation; it also permits effective analgesia and sedation to be administered and keeps the infant immobile to protect the wound and the TAT tube. Because the gut is extensively handled, a paralytic ileus is expected and the infant must remain on the ‘drip and suck’ mode of management until some motility returns. Once gut motility has commenced, small enteral feeds, ideally of expressed breast milk, may be commenced. Supporting the mother to express her breast milk and ensuring the future nutrition of the infant in line with World Health Organisation and Department of Health recommendations is a key children’s nurse role and the RCN best practice guidelines (2009) can help inform unit policy. Typically, these children are quickly extubated and can be moved to a high dependency area or a cubicle in a children’s ward. As it can sometimes take these infants a while to tolerate full feeds, full or fractional PN is maintained via a long line while full enteral feeding is established. This can be established on the children’s ward, however this can be a frustrating time for the family as the infant is otherwise well and handles normally. Reverse transfers to the referring hospital may relieve some for the pressure on the family, however clear PN prescription and feeding re-grade plans have to be in place and suitable follow-up organised.
Colonic Atresia
This is rare but life-threatening if left untreated. In colonic atresia, the problems are complete bowel obstruction through which gas and stool cannot pass, the colonic segment above the atresia becomes distended and, if left untreated, leads to perforation (Nielson and Zitsman 2008). Davenport and colleagues (1990) reviewed the incidence of colonic atresia and suggest the following incidence within their sample of population.
- Ascending colon – 28%.
- Transverse colon – 23%.
- Splenic flexure – 25%.
- Descending and sigmoid – 20%.
- Hepatic flexure – 3%.
Treatment
A surgical repair with resection of the atretic component and end-to-end anastomosis is required. In complex cases where there may be multiple atresias or where the repair has been difficult the formation of a temporary stoma may be employed to protect the anastomosis. General postoperative care principles are required to manage the infant, with consideration given to fluid management, electrolyte balance, pain management and the reinstitution of enteral feeding when clinically indicated.
Colonic Stenosis
Narrowing of the colon is much more common and frequently occurs following necrotising enterocolitis. In colonic stenosis, the problem is that gas and stool can pass through a narrow area and while the infant is passing milk stools, this may not be noticeable. When the diet changes from breast milk or formula to cereals and solid foods, the stool becomes more formed. This will result in the stenosis becoming symptomatic. The toddler can present with signs of obstruction, distension, feeding intolerance or faltering growth (Nielson and Zitsman 2008).
Gastroschisis
This occurs due to a congenital anterior abdominal wall defect usually to the right of the umbilicus which results in the evisceration of abdominal contents. The incidence of this condition has been increasing, however this may not be a year-on-year trend (Office for National Statistics 2006, 2007) and the risk is not geographically equally distributed (Kilby 2006), with more infants with this defect being born in the north of England and to younger mothers. Advances in neonatal care, total parenteral nutrition, surgical confidence and technique have all improved the prognosis and 90% of affected babies now survive. Long-term prognosis is also positive as late mortality following the infant year is rare and usually unrelated to the primary defect (Davies and Stringer 1997). Complications from the original defect requiring prolonged hospital management or further corrective intervention may result from the condition of the bowel at birth, prolonged paralytic ileus, malrotation, malabsorption, adhesions or abnormal bowel habit.
Diagnosis is usually made antenatally following foetal scan and there seems to be no difference in outcome if the infant is delivered by caesarean section or vaginally. The aim, once the infant is delivered, is to keep the exposed bowel moist and the infant should be inserted feet first into a clear plastic sac. This method is preferred over packs soaked with saline, although these can be used in an emergency. The bag should be tied above the lesion leaving the arms free so the infant can be cannulated for IV access, as to the circulation using a long line, as TPN and a range of broad spectrum antibiotics will be required. The infant needs to be nursed supported on their side to prevent kinking the loops of bowel and transferred once stable to the surgical unit. If the infant is self-ventilating with ease and maintaining their oxygen saturations, there is no need to intubate for transfer as prolonged ventilation is damaging to newborn lungs. This infant will be intubated and ventilated for surgery and the immediate recovery period to address the requirement for suitable analgesia and for ease of nursing to ensure that the infant is kept immobile to prevent tension on the repair line.
Repairs may be primary or staged depending on the extent of the defect. If the repair is staged some sort of silo bag to contain the gut will be stitched to the abdomen. There is a variety of makes, e.g. Gore-Tex®, or the plastic from an infusion bag may be used. This silo is created to accommodate intestines and will be reduced over a period of time. This avoids creating excessive intra-abdominal pressure, allows skin to stretch and grow, and improves the cosmetic result. The bag containing the intestines needs to be supported or suspended from the incubator roof or a frame to avoid the bag containing the gut from folding over causing trauma or kinking of the bowel. A combination of the primary defect and handling the gut normally results in a paralytic ileus and these infants are usually nil by mouth for prolonged periods until peristalsis returns. The mother should be supported and strongly encouraged to express and store her breast milk as this is the preferred feed when feeding is eventually commenced. Building up feed tolerance can be a frustrating time for the parents as by this time the defect has been repaired the wound has healed and their baby is dressed and looks ‘normal’. Reverse transfers to the referring hospital may relieve some for the pressure on the family, however clear feeding re-grade plans have to be in place and suitable follow-up organised.
Exomphalos or Large Omphalocoele
This is another abdominal wall defect where the gut has herniated into the base of the umbilical cord. It usually looks less drastic than the gastroschisis but the prognosis can be poorer as there is quite a strong association between this and other midline defects and some of these are chromosomal in origin. Not all of these are surgically repaired as some of the smaller ones may epithelialise over.
Intussusception
This is the telescoping of one segment of intestine into the adjacent distal segment which compresses the attached associated mesentery, blood vessels and nerves as a consequence. The resulting oedema and ischaemia to the affected intestine leads to obstruction and the risk of perforation and peritonitis. Most cases affect the ileo-colic region and it is the most common cause of intestinal obstruction in children between 3 months and 6 years of age, but small abnormalities can also provide a focus for the telescoping in clinical conditions such as a Meckel’s diverticulum (Milbrandt and Sigalet 2008). Intussusception is rare in infants under 3 months of age and in older children and young people. Intussusception may also be a rare postoperative complication occurring in 0.08–0.5% of laparotomies. The likely mechanism is due to a difference in activity between segments of the intestine recovering from an ileus, which produces the intussusception (Bai et al. 2009).
Signs and Symptoms of Intussusception
- Colicky abdominal pain.
- Vomiting, feed intolerance; the vomit may contain bile.
- Stools containing blood called ‘currant jelly’ stool.
- Tubular mass in the abdomen.
- Absent bowel sounds.
- Abdominal X-ray may reveal gas and or free fluid levels.
If the infant is not managed in a timely fashion with appropriate fluid resuscitation, the condition can escalate and result in shock, with the infant requiring full intensive care provision, including inotropic support.
Management of Intussusception
If the infant is sufficiently well, a non-surgical intervention can be tried with the administration of either an air enema or a barium contrast enema, which will confirm the diagnosis and can also rectify the problem. The enema itself carries a risk of perforation and cannot be performed if there is a suspicion that the bowel has already perforated (Shekherdimian et al. 2009).
If the intestinal obstruction cannot be resolved by the enema, surgery is necessary to reverse the intussusception and relieve the obstruction. This can sometimes result in resection and anastomosis to remove the segment of the intestine which has become gangrenous. This procedure may be undertaken laparoscopically if there is an experienced practitioner to perform the procedure (Fraser et al. 2009).
Volvulus or Malrotation
Volvulus can be defined as a twisting of a loop of intestine around its mesenteric attachment site. This twisting can occur at various sites of the GI tract, including the stomach, small intestine, caecum, transverse colon and sigmoid colon. Midgut volvulus refers to twisting of the entire midgut around the axis of the superior mesenteric artery, which can be a very serious presentation. Because of embryological maldevelopment and rotation the condition can present soon after birth or may manifest within two months of life. It can be associated with a number of other pathologies, such as duodenal atresia, Meckel’s diverticulum, intussusception, small bowel atresia, prune belly syndrome, gastric volvulus, persistent cloaca, Hirschsprung disease and extrahepatic biliary anomalies.
Clinical Presentation of Volvulus
The acute presentation resembles an abdominal emergency and requires urgent intervention:
- Pain and tender abdomen.
- Vomiting, which is usually bilious.
- Infants or children with delayed diagnosis may present in a shocked state.
Subacute presentation, usually found in older children, is not such an urgent situation and is not usually seen in intensive care. These children may have abdominal pain which is disproportionate to abdominal distension and may have a palpable mass. Abdominal X-rays may demonstrate small bowel obstruction with fluid–air levels, while contrast studies may demonstrate corkscrew signs.
Management of Volvulus
- Management of pain.
- Gastric decompression with NGT placement and left on free drainage.
- Fluid and electrolyte replacement.
- Intravenous antibiotics.
Acute or shocked presentation necessitates early surgery to prevent the development of ischaemia and or gangrenous bowel segments. Most children will have a Ladd procedure where the appendix is removed, the mesenteric bands are divided and the small intestine is replaced to the right and the colon to the left side of the abdominal cavity.
Postoperative Management
Standard postoperative care will be required. Some children with volvulus suffer with prolonged ileus postoperatively and will require parenteral nutrition as a consequence of being unable to tolerate enteral nutrition. These children will require gentle reintroduction of enteral feeding, usually determined by the surgical team. A very small number will develop acquired short bowel syndrome and will require ongoing management.
Abdominal Trauma
Abdominal trauma is relatively uncommon in children and most are managed conservatively regardless of age (Gaines 2009; Tataria et al. 2007). Most abdominal trauma results from blunt force trauma. In isolation it has a low mortality but when combined with other trauma, such as neurological or thoracic, the mortality rises (Davenport and Pierro 2009). Young children’s abdominal organs are vulnerable as the ribs are poorly ossified and significant injuries in the under 2 year olds should trigger ‘safeguarding questions’ as there may be non-accidental, welfare and supervision issues (Champion et al. 2002).
Complications of Major Organ Lacerations
- Major haemorrhage.
- Intestinal perforation.
- Leakage of gastrointestinal secretions into the abdominal cavity.
Abdominal organs such as the liver and spleen are highly vascular and friable so they are susceptible to laceration. These injuries can be life-threatening. If suspected, the child should be urgently admitted to a PICU, as the levels of medical expertise in Emergency Departments are variable (Prentiss and Vinci 2009).The child needs circulatory access, should be fully monitored and a range of diagnostic investigations performed. There is still a role for plain abdominal X-rays, but ultrasound and a CT scan can provide a more detailed picture. As considerable amounts of blood can be lost into the abdomen with only minor increases in girth size, measuring girth is an inaccurate means of observation (Atkin and Clifford 1985) yet continues to be performed in some units. This can lead to excessive handling and increase the distress of the child. Perforation of the gut can be a complication of blunt trauma and spillage of gut contents may lead to peritonitis. This is regarded as secondary peritonitis.
The aims of medical treatment and surgical management are twofold and complementary. One focuses on the need to restore and maintain the circulatory volume and the other to locate the area(s) of damage and prevent further blood loss. If surgery is indicated, then primary repair is preferred, but when not possible excision of the affected lobe of liver or removal of the spleen may be required. The liver has an amazing capacity to regenerate but there are considerable immunity risks for the asplenic child. There is some controversy with regard to the need for prophylaxis antibiotic therapy post-splenectomy (Moffatt 2009) and practitioners should refer to their local policies in terms of treatment strategies and medications utilised.
Neonatal Liver Disease
In neonates, jaundice can occur as a feature unrelated to a primary liver disorder or a symptom exacerbated by coexisting diseases. It is considered here as neonates frequently get admitted to the PICU for surgery or when the neonatal unit is full. It is common among premature infants and indeed in some term babies. The speed in which jaundice occurs is significant and also the length of time the condition is present is important as these can be indicators of the general well-being of the infant. Generally, physiological jaundice appears by day 3 and with good management lasts about 2 weeks. The causes of neonatal jaundice can be split into two main groups: haemolytic, caused by increased red cell breakdown; and non-haemolytic, caused by impairment in bilirubin excretion, as in liver disease. Haemolytic hyperbilirubinaemia is usually associated with unconjugated or indirect bilirubin.
Unconjugated Hyperbilirubinaemia
This accounts for most cases of jaundice. The neonatal liver is fairly immature and bilirubin is the major waste product of haemoglobin degradation, which mainly takes place in the spleen. A raised bilirubin level in the blood is called hyperbilirubinaemia. This spills out of the bloodstream into the tissues and causes the yellow discoloration associated with the condition. The main causes are listed in Table 8.6.
Table 8.6 Causes of unconjugated hyperbilirubinaemia
Source: Dixon et al. (2009) Nursing the Highly Dependent Child or Infant: A Manual of Care. Reproduced with permission from John Wiley and Sons, Ltd.
| Cause | Aetiology |
| Physiological jaundice | Due to excessive RBC breakdown at birth and functional immaturity of the conjugation processes in the liver. |
| Term babies treated with phototherapy if bilirubin greater than 300 µmol/l. | |
| Breast milk jaundice | Persistence of foetal mechanisms (e.g. beta glucuronidase) causes low-level continued jaundice in some infants. This can be exacerbated by poor feeding techniques leading to dehydration. Surveillance is required by doctor if it lasts over a month. |
| Systemic disease | |
| Rhesus and ABO incompatibility Glucose-6-phosphate dehydrogenase Sickle cell disease Thalassaemia Spherocytosis Hypothyroidism Upper-intestinal obstruction Sepsis Hypoxia/acidosis Galactosaemia Fructosaemia | All cause increased haemolysis, which is particularly problematic if encountered in the newborn period exacerbating jaundice levels because of liver immaturity. |
| Iatrogenic | |
| Trauma at birth Blood transfusion Administration of certain drugs | Bruising leading to breakdown of RBC. Administration process can damage RBC. Some drugs displace bilirubin from albumin so increasing the free component, e.g. diazepam, hydrocortisone, gentamicin, cefalosporins, digoxin. |
| Inherited disorders | |
| Crigler-Najjar syndrome (Types 1 and 2) | Deficiency in enzyme uridinediphosphateglucuronosyltransferase (UDPGT). Type 1 is total and needs phototherapy long term. Type 2 is partial and may ultimately be treated with enzyme inducers such as phenobarbitone. |
| Gilbert’s syndrome | Mild, transient jaundice that needs no treatment but reassurance. |
Management
Low-level unconjugated hyperbilirubinaemia in the neonate does not need active intervention other than to establish a means of effective enteral feeding to improve hydration and stimulate gut transit times. Blood is sampled for levels of bilirubin to be measured and plotted on a chart which indicates treatment thresholds. If the infants are very sick, these treatment thresholds may be lowered as these infants may be considered to be more vulnerable to the consequences of kernicterus. Usually, treatment is by phototherapy but extremely high levels may be managed by exchange blood transfusion.
Providing Phototherapy in the PICU
Phototherapy works by using light to change bilirubin to the more water-soluble cis-form, which is easier to excrete. A range of lights, pads and blankets is now used. However, for phototherapy to be effective, the infant requires maximum skin exposure and in some cases this means being naked except for a nappy. This can cause some parents distress so attention to the environment of care using screens, turning the baby round so they are head first visible rather than bottom first, or nursing the infant at the end of the unit away from the main unit walkway.
- Maintaining maximum exposure also has implication for handling and parents keen to get to know their new baby can feel excluded.
- Sick infants can have a critically unstable temperature which exposure to phototherapy exacerbates. Central and shin temperatures should be monitored continually.
- The infant’s eyes must be covered when under lights, again causing some distress to the parents, and constantly monitored to ensure the covers remain in situ. Eye care should be provided when the nappy is changed and the parents can be taught to do this.
- Skin care is important to remove acidic urine and stools, however skin creams, oils or ointment must be avoided as the heat produced by the lights may result in skin burns.
Nurses play an important part in supporting the family to come to terms with this additional complication. In the NNU phototherapy is more common and as parents see other infants having it without difficulty they are more accepting. In the PICU their infant may be the only one in a box or under a set of lamps, so the distress may be disproportional.
If exchange transfusion is considered a textbook on neonatal care should be consulted.
Conjugated Hyperbilirubinaemia
This is much less common. Elevated conjugated bilirubin results from obstructive liver disease, but can be seen in many types of liver disease which affect hepatocyte function. Conjugated hyperbilirubinaemia is said to be present when >20% of the total plasma bilirubin is conjugated or when the total conjugated level is greater than 20 mmol/l. Total and conjugated bilirubin levels are measured in the laboratory and the level of unconjugated is the difference between the two. The ratios of types of bilirubin can be helpful in diagnosis of the cause. In hepatobiliary disease the total and conjugated will be raised. Total and unconjugated will be raised in hepatic disease. A split bilirubin (total and conjugated) should be performed on any baby who remains jaundiced after 2 weeks of life (3 weeks for preterm infants). If the conjugated fraction is raised as defined above, investigations for possible liver disease should be started. Liver disease in the newborn can present as:
- An ill infant with liver failure (deranged clotting unresponsive to intravenous vitamin K).
- Neonatal hepatitis syndrome.
- Biliary obstruction.
If an infant has pale stools and dark urine, this needs to be brought to the attention of the medical team urgently. These infants are going to require more investigation for their hyperbilirubinaemia, and management and treatment will be more complex as phototherapy is not indicated for this group of babies.
Biliary Atresia
This is one of the most common reasons for liver transplantation in children. The cause of this remains relatively obscure and because it is rare occurring in 1/15 000 (Davenport and Pierro 2009) there is little research to inform on the aetiology or epidemiology of the condition. Biliary atresia is a condition in which the normal extrahepatic biliary system is disrupted. Progressive damage of extrahepatic and intrahepatic bile ducts secondary to inflammation may occur, leading to fibrosis, biliary cirrhosis and eventual liver failure.
There are three main types of biliary atresia:
- Type I: the common bile duct is obliterated, while the proximal bile ducts are patent.
- Type II: atresia of the hepatic duct is seen, with cystic bile ducts found at the porta hepatis.
- In type IIa, the cystic and common bile ducts are patent.
- In type IIb, the cystic, common bile duct, and hepatic ducts are obliterated.
- In type IIa, the cystic and common bile ducts are patent.
- Type III: discontinuity of the right and the left hepatic ducts to the level of the porta hepatis. This form of biliary atresia is common, accounting for more than 90% of cases.
The infant with biliary atresia usually appears normal at birth but develops jaundice 2–3 weeks after birth. The key symptoms are:
- Jaundice.
- Dark urine: build-up of bilirubin.
- Acholic (clay-coloured) stools.
- Hepatomegaly.
- Weight loss and irritability.
Management of Biliary Atresia
Surgical intervention, known as a hepatoportoenterostomy or a Kasai procedure, is the only management option for biliary atresia and is ideally undertaken before 60 days of life to limit scarring within the liver, but this is not a curative procedure and the majority of children with biliary atresia will require liver transplantation before they reach adulthood (Bassett and Murray 2008).
The Kasai procedure removes the abnormal bile ducts and a loop of intestine is mobilised and sutured to some of the smaller ducts to drain the liver. As a result, bile flows from the smaller bile ducts straight into the intestine, bypassing the need for the larger bile ducts completely. This reduces reabsorption of bile into the blood stream and ensures that digestion of enteral nutrients is supported. This corrects many of the problems of biliary atresia.
Cholangiopathies
Cholangiopathies are a wide array of congenital or acquired disorders that result in chronic cholestatic conditions which lead to liver failure (Lazaridis et al. 2004). Cholangiopathies share the common feature of primarily targeting cholangiocytes, the epithelial cells lining the intrahepatic biliary tree (Lazaridis et al. 2004). These diseases are characterised by the progressive vanishing of bile ducts (ductopenia) which results from an abnormal cholangiocyte homeostasis. It is thought that this ductopenia results from excessive cell death by apoptosis (Alvaro et al. 2007). There is a compensatory proliferative response which enlarges the liver and is one reason why these children have large swollen abdomens. Cholangiopathies are a challenge for clinicians to manage and 50% of transplants among paediatric patients are due to these disorders.
Acute Liver Failure (ALF)
Fulminant hepatic failure or acute liver failure (ALF) in children is a relatively rare clinical syndrome and the mortality rate is high at 60–80% in the absence of liver transplantation (Nazer 2011, Cochran and Losek 2007). The complexity of the child with hepatic failure is a challenge for the whole healthcare team. This is not the same as the transient liver dysfunction associated with critical illness which is commonly seen in PICU, however critical illness with persistent end organ ischaemia can be a contributory factor.
The three essentials for normal liver function are adequate blood flow, good oxygenation and low pressure in the biliary system. The functions of the liver are numerous but include the following:
- Carbohydrate, fat and protein metabolism.
- Removal of drugs and hormones.
- Excretion of bile and the synthesis of bile salts.
- Storage of glycogen.
- Activation of vitamin D, and synthesis of vitamins A, B12, D, E and K.
- Production of apoferetin.
- Phagocytosis.
The causes of acute or fulminant liver failure are wide-ranging. The mechanisms of hepatic cellular injury which may lead to ALF are:
- Direct hepatocellular injury:
- Herpes virus family.
- Toxic or reactive metabolites (e.g. paracetamol).
- Toxic metabolites of compounds – metabolic diseases.
- Herpes virus family.
- Immune-mediated hepatocellular injury:
- Viral infections.
- Drug hepatotoxicity (dihydralazine, halothane).
- Viral infections.
- Ischaemic hepatocellular injury:
- Shock states
- Systemic inflammatory response syndrome (SIRS).
- Shock states
Clinical Features of ALF
Acute liver failure can develop within days or weeks depending on the underlying aetiology. The range of clinical signs will also vary; however there is a rapid onset of hepatic dysfunction with associated development of coagulopathies, metabolic derangement and the accumulation of neurotoxic by-products in the brain which cause an encephalopathic picture to emerge.
General Signs of ALF
- Nausea and vomiting.
- Fatigue.
- Anorexia.
- Jaundice.
- Ascites.
- Increased bruising.
Hepatic Signs of ALF
- Elevated transaminases (ALT, AST).
- Hypoglycaemia.
- Abnormally low cholesterol levels.
- Coagulopathy (not correctable with parenteral vitamin K).
- Progressively rising bilirubin.
- Elevated ammonia levels with associated encephalopathy.
Hepatic Encephalopathy (HE)
HE is a gradable, reversible syndrome characterised by decreased level of consciousness, seizures or multifocal muscle twitching or coma. For HE to be diagnosed there must be an absence of other factors which could suppress cerebral function. Up to 80% of patients with HE have cerebral (cytotoxic) oedema while between 30–50% will have clinically significant raised intracranial pressure.
HE may be graded according to the neurological signs present (Table 8.7).
Table 8.7 Grading of hepatic encephalopathy
Source: adapted from Arya, SheffaliGulati and Deopujari 2010; Blei and Córdoba 2001; Munoz 2008.
| Grade of encephalopathy | Clinical features |
| Grade I (Mild) | Lethargy, disruption of day–night sleep patterns, mild motor impairment. |
| Grade II (Moderate) | Disorientation, confusion, inappropriate behaviours, increasing drowsiness but remaining responsive to simple commands. |
| Grade III (Severe) | Rousable to voice and localisation to pain, confusion, incoherent speech patterns. |
| Grade IV (Coma) | Unrousable, minimal response to painful stimuli, decerebrate or decorticate posturing. |
Management of ALF
The management of ALF is determined in part by the need to manage the complications associated with the hepatic dysfunction alongside therapies aimed at minimising ongoing hepatic cell damage and supporting the child until there is recovery of hepatic function or the child is referred for transplant (Table 8.8).
Table 8.8 Management strategies for ALF
Source: Arya et al. 2010; Blei and Córdoba 2001; Latif and Mehmood 2010; Munoz 2008.
| Body system | Management strategies |
| Cardiovascular | Restoration of a euvolaemic state with judicious use of fluid and CVP monitoring. |
| Diuresis with albumin, fluid restriction, diuretics. Support myocardial performance – use of vasoactive medications. Maintain good oxygenation to the myocardium. | |
| Pulmonary | Intubation and mechanical ventilation may be necessary, particularly in children with grade III/–IV HE and in these children, oral intubation is indicated to prevent potential haemorrhage secondary to coagulopathy. |
| Reduce the risk of ventilator associated pneumonia (Chapter 4). | |
| Neurological | ICP monitoring is controversial and not proven to benefit the outcome but could be considered in children with rapidly developing signs and symptoms of raised ICP, Grade IV HE, rapidly progressing grade III HE or the presence of cerebral oedema on CT scan. |
| Standard RICP therapy to maintain ICP <20 mmHg and CPP >50 mmHg (Chapter 7). | |
| Barbiturate coma may be used if necessary. | |
| Continuous EEG monitoring. | |
| Continuous renal replacement therapy (CRRT) such as continuous venovenous haemofiltration (CVVH) or plasmapheresis to reduce the levels of harmful nitrogenous metabolites. | |
| Metabolic | Nil by mouth in initial stages until cause identified. |
| Maintain serum blood glucose level above 4 mmol to minimise the possibility of hypoglycaemia and its deleterious effects. | |
| Maintain glucose intake with 15, 20, or 50% glucose as necessary. | |
| Enteral feeds/dietary intake which is low in sodium and protein (if stable – grade I–II HE). | |
| Use of PN – highest concentration of glucose tolerated if unable to maintain enteral nutrition with supplements of amino acid (trophamine) 0.5–1.0 g/kg/day and the use of lipids (20% solution) 0.5–3 g/kg/day. | |
| Haematological | Vitamin K – 0.2 mg/kg/day (max. 10 mg) IV × 3 days, then every other day. |
| Maintain PT at 20–25 sec (if no active bleeding). | |
| Maintain PT at <20 sec (if active bleeding). | |
| Maintain platelet count >50 000. | |
| Maintain haematocrit > 30%. | |
| Fresh frozen plasma (FFP) infusions for active bleeding. | |
| Plasmapheresis should be considered when there is severe coagulopathy and/or bleeding. | |
| Gastrointestinal | Gastric protection to prevent ulceration and reduce the risk of gastric bleeding – H2 receptor antagonist. |
| Use of lactulose to reduce the transit times of ammonia containing matter through the bowel. | |
| Renal | Conventional renal support through the use of peritoneal dialysis or CVVH may be required. |
| Renal dysfunction with renal failure occurs in as many as 50% of patients. The kidneys are involved secondary to hepatorenal syndrome (HRS), acute tubular necrosis (ATN), drug-induced nephrotoxicity or prerenal azotaemia. | |
| Hepatorenal syndrome | |
| HRS is defined as functional renal failure occurring in patients with severe liver disease in the absence of any other underlying cause of renal disease. A decrease in blood flow to the kidneys has been suggested as the underlying pathophysiology (Latif and Mehmood 2010). | |
| Liver transplantation is the treatment of choice for HRS; however, some patients continue to require dialysis even following the transplant. |
Complications arising from hepatic dysfunction include those detailed below. The most common causes of death are notated with an asterisk:
- Encephalopathy.
- Cerebral oedema and raised intracranial pressure.*
- Haemorrhage.
- Hypoglycaemia (profound).
- Disseminated intravascular coagulopathy.*
- Acid base derangements.
- Cardiac and circulatory instability.
- Pulmonary failure.
- Renal failure.
- MODS.*
- Sepsis.*
Bridging to Transplant or Recovery
Referral to a specialist centre is necessary for any child who may require liver transplantation. These centres offer a range of bridging therapies and not all children will proceed to a full transplant. Current strategies include hepatocyte transplantation or extracorporeal liver support systems. These are highly specialised therapies and practitioners should seek further information from specialist centres.
Auxiliary Orthotopic Liver Transplantation (AOLT)
AOLT usually involves the transplanting of one or two lobes of the donor liver, although the whole liver may be used without the complete removal of the native liver. AOLT acts as a bridge to native liver recovery. If recovery occurs, then immunosuppression may be discontinued and the graft removed or allowed to atrophy to avoid long-term immunosuppression. In metabolic diseases, AOLT can provide the necessary enzymes to correct the deficiency and yet allows the patient to continue to utilise their native liver. Published studies indicate a good success rate for this therapeutic approach (Faraj et al. 2010).
Predictive Outcomes of ALF
The overall survival for patients with ALF and grade III or grade IV HE has increased from 20% to 50% over the past two decades without transplant, but the best independent predictor of outcome is the aetiology of the ALF. For example, Wilson’s disease has a predicted survival of <5% without liver transplant while the survival from non-ABC hepatitis is <20%.
Liver Transplant
Paediatric liver transplantation is highly successful and is now an established therapeutic option (Alagille 2004).Children who have severe hepatic disease and who may require a transplant are referred via established care pathways to a paediatric liver transplant centre. There are many reasons why a child may need to be considered for transplant and Table 8.9 is not an exhaustive review. As children’s size and age varies widely, matched grafts are unlikely to be available. This has led to the development of reduced-size graft and split liver transplantation. This has the advantage that a single donor liver can serve two or three children. Parents or adult relatives may also be screened and selected to provide a segment of their liver in living donor liver transplantation. Post-transplant management protocols usually include the use of a calcineurin inhibitor (cyclosporine or tacrolimus) and while the protocol usually depends on the surgical team’s preference, tacrolimus has almost completely replaced cyclosporine in liver transplantations (Jain et al. 2002). Most centres also use steroids for some time post-transplant.
Table 8.9 Indications for liver transplant
Source: UK Transplant 2005.
| Life-threatening metabolic liver disease | Crigler–Najjar syndrome |
| Urea cycle defects | |
| Hypercholesterolaemia | |
| Organic acidaemias | |
| Primary hyperoxaluria | |
| Chronic liver disease | Biliary atresia |
| Alpha-1-antitrypsin deficiency | |
| Autoimmune hepatitis | |
| Sclerosing cholangitis | |
| Caroli syndrome | |
| Wilson’s disease | |
| Cystic fibrosis | |
| Progressive familial intrahepatic cholestasis (all types) | |
| Alagille syndrome | |
| Glycogen storage diseases types 3 and 4 | |
| Tyrosinaemia type 1 | |
| Acute liver failure | Ingestion of toxic metabolites |
| Liver tumours | Unresectable hepatoblastoma (without active extrahepatic disease) |
| Unresectable benign liver tumours with disabling symptoms |
Endocrine Presentations
There are a small number of endocrine presentations where the child may either in the first instance or as a secondary consequence end up in the intensive care setting. Endocrinology is a specialised field and the general principles of management only are outlined in this section.
Diabetic Ketoacidosis
Diabetes mellitus (DM) arises from inadequate insulin effects within the body. In children it almost always occurs as a result of inadequate amounts of insulin being available.
TYPES OF DM
- Type I (IDDM): the most frequent form in children.
- Type II (NIDDM): uncommon in children, however the incidence is increasing (these children are not usually prone to ketosis).
RARE TYPES OF DM
- Genetic defects of β-cell function.
- Genetic defects of insulin action.
- Diseases of the exocrine pancreas.
- Drug or chemical induced (Table 8.4).
- Uncommon forms of immune-mediated diabetes.
- Other genetic syndromes (Alberti et al. 1999).
The mechanism for the development of DM in children is not clearly understood but there are a number of possible factors which may predispose a child towards it, including genetic predisposition, following a viral infection or as part of an autoimmune process. A number of children have either defective conversion of pro-insulin to insulin or an abnormal structure of their insulin molecules which causes the development of DM.
Diabetic Ketoacidosis
DKA is one of the commonest metabolic disorders seen in the paediatric population and while the majority of children will not be managed in the intensive care setting, a small number will due to their age or persistent metabolic derangement which is slower than usual to respond to treatment strategies.
Pathophysiology of Diabetic Ketoacidosis
DKA is characterised by severe depletion of water and electrolytes from both the intracellular (ICF) and extracellular fluid (ECF) compartments. The initial and key event in DKA is insulin deficiency, however many of physiological processes seen are mediated by increased levels of counter-regulatory hormones (glucagon, adrenaline and noradrenaline, growth hormone and/or cortisol) (Table 8.10).
Table 8.10 Physiological changes of insulin and counter-regulatory hormones in DKA
| Physiological effects | Changes in DKA | |
| Insulin | Helps glucose enter the cells. Stimulates glycogenesis (creation of glycogen). Stimulates glucose catabolism (breakdown). Lowers serum blood glucose level. | Decreased or absent levels mean that glucose does not enter the cells. Glycogen is not formed to store glucose and therefore the serum level of glucose rises. |
| Cortisol | Stimulates protein mobilisation and gluconeogenesis. | Increased levels of proteins, which are utilised as an alternative source of energy. |
| Raises the serum blood glucose level. | ||
| Glucagon | Stimulates glycogenolysis (liberation of glycogen from the liver stores). Raises the serum blood glucose level. | Increased circulating levels of glycogen, which is metabolised to glucose but which cannot be used by the cells for energy. |
| Growth hormone | Decreases utilisation of carbohydrate for energy. | Increased levels of fat, which is broken down into glycerol and fatty acids – neither can be accessed by cells for energy. |
| Stimulates the liberation and catabolism of fats. | ||
| Raises the serum blood glucose level. | ||
| Adrenaline | Stimulates the liberation and catabolism of fats. Stimulates glycogenolysis (liberation of glycogen from the liver stores). Raises the serum blood glucose level. | Increased levels of fat which is broken down into glycerol and fatty acids – neither can be accessed by cells for energy. Increased circulating levels of glycogen, which is metabolised to glucose but which cannot be used by the cells for energy. |
As a consequence of the processes outlined in Table 8.10 the five key clinical features of DKA are hyperglycaemia, ketogenesis, hyperosmolality, dehydration and electrolyte imbalance (Figure 8.1). The key to the management of the child with DKA is thorough assessment followed by frequent reassessment of the therapies instituted. Treatment is divided into two phases: initial assessment and management on presentation; and long-term management. (For further information regarding pre-intensive care management, practitioners are advised to refer to the current British Society of Paediatric Endocrinology and Diabetes (BSPED) guidelines and local integrated care pathways.)
Figure 8.1 The pathways for the development of the clinical features of DKA.
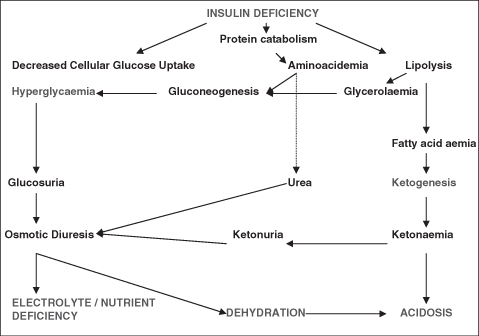
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


