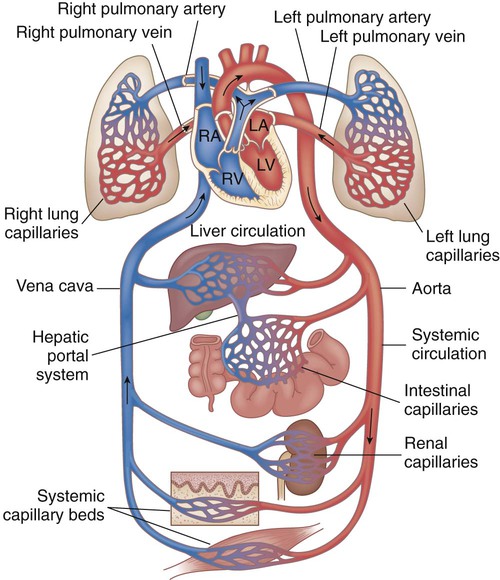
Chapter 42 On completion of this chapter, the reader will be able to: • Design a plan for assisting children during cardiac diagnostic procedures. • Demonstrate an understanding of the hemodynamics, distinctive manifestations, and therapeutic management of congenital heart disease. • Outline a care plan for an infant or child with heart failure. • Describe the care for a child who has hypoxia. • Describe the care for an infant or a child with a congenital heart defect and its surgical repair. • Discuss the nurse’s role in helping the child and family cope with congenital heart disease. • Differentiate between rheumatic fever and rheumatic heart disease. • List the criteria for selected cholesterol screening of children. • Discuss the assessment and management of hypertension in children and adolescents. • Outline a care plan for a child with Kawasaki disease. • Describe the emergency treatment for shock, including anaphylaxis. Cardiovascular disorders in children are divided into two major groups—congenital heart disease and acquired heart disorders. Congenital heart disease (CHD) includes primarily anatomic abnormalities present at birth that result in abnormal cardiac function. The clinical consequences of congenital heart defects fall into two broad categories—heart failure (HF) and hypoxemia. Acquired cardiac disorders are disease processes or abnormalities that occur after birth and can be seen in the normal heart or in the presence of congenital heart defects. They result from various factors, including infection, autoimmune responses, environmental factors, and familial tendencies. The pathophysiology review found in Fig. 42-1 describes the flow of blood through the heart. The physical assessment of suspected cardiac disease begins with observation of general appearance and then proceeds with more specific observations. The following are supplementary to the general assessment techniques described for physical examination of the chest and heart in Chapter 29: Nutritional state—Failure to thrive or poor weight gain is associated with heart disease. Color—Cyanosis is a common feature of CHD, and pallor is associated with poor perfusion. Chest deformities—An enlarged heart sometimes distorts the chest configuration. Unusual pulsations—Visible pulsations of the neck veins are seen in some patients. Respiratory excursion—This refers to the ease or difficulty of respiration (e.g., tachypnea, dyspnea, expiratory grunt). Diagnostic catheterizations—These studies are used to diagnose congenital cardiac defects, particularly in symptomatic infants and before surgical repair. They are divided into (1) right-sided catheterizations, in which the catheter is introduced through a vein (usually the femoral vein) and threaded to the right atrium (most common); and (2) left-sided catheterizations, in which the catheter is threaded through an artery into the aorta and into the heart. Interventional catheterizations (therapeutic catheterizations)—A balloon catheter or other device is used to alter the cardiac anatomy. Examples include dilating stenotic valves or vessels or closing abnormal connections. Electrophysiology studies—Catheters with tiny electrodes that record the impulses of the heart directly from the conduction system are used to evaluate dysrhythmias and sometimes destroy accessory pathways that cause some tachydysrhythmias. Cardiac catheterization has become a routine diagnostic procedure and may be done on an outpatient basis. However, it is not without risks, especially in neonates and seriously ill infants and children. Possible complications include acute hemorrhage from the entry site (more likely with interventional procedures because larger catheters are used), low-grade fever, nausea, vomiting, loss of pulse in the catheterized extremity (usually transient, resulting from a clot, hematoma, or intimal tear), and transient dysrhythmias (generally catheter induced) (Uzark, 2001). Rare risks include stroke, seizures, tamponade, and death. Preparing the child and family for the procedure is the joint responsibility of the patient care team. School-age children and adolescents benefit from a description of the catheterization laboratory and a chronologic explanation of the procedure, emphasizing what they will see, feel, and hear. Older children and adolescents may bring earphones and favorite music so they can listen during the catheterization procedure. Preparation materials such as picture books, videotapes, or tours of the catheterization laboratory may be helpful. Preparation should be geared to the child’s developmental level. The child’s caregivers often benefit from the same explanations. Additional information, such as the expected length of the catheterization, description of the child’s appearance after catheterization, and usual postprocedure care, should be outlined. (See also Prepare the Child and Family for Invasive Procedures, p. 1341.) • Pulses, especially below the catheterization site, for equality and symmetry (Pulse distal to the site may be weaker for the first few hours after catheterization but should gradually increase in strength.) • Temperature and color of the affected extremity, because coolness or blanching may indicate arterial obstruction • Vital signs, which are taken as frequently as every 15 minutes, with special emphasis on heart rate, which is counted for 1 full minute for evidence of dysrhythmias or bradycardia • Blood pressure (BP), especially for hypotension, which may indicate hemorrhage from cardiac perforation or bleeding at the site of initial catheterization • Dressing, for evidence of bleeding or hematoma formation in the femoral or antecubital area • Fluid intake, both IV and oral, to ensure adequate hydration (Blood loss in the catheterization laboratory, the child’s NPO status, and diuretic actions of dyes used during the procedure put children at risk for hypovolemia and dehydration.) • Blood glucose levels, for hypoglycemia, especially in infants, who should receive dextrose-containing IV fluids Depending on hospital policy, the child may be kept in bed with the affected extremity maintained straight for 4 to 6 hours after venous catheterization and 6 to 8 hours after arterial catheterization to facilitate healing of the cannulated vessel. If younger children have difficulty complying, they can be held in the parent’s lap with the leg maintained in the correct position. The child’s usual diet can be resumed as soon as tolerated, beginning with sips of clear liquids and advancing as the condition allows. The child is encouraged to void to clear the contrast material from the blood. Generally, there is only slight discomfort at the percutaneous site. To prevent infection, the catheterization area is protected from possible contamination. If the child wears diapers, the dressing can be kept dry by covering it with a piece of plastic film and sealing the edges of the film to the skin with tape. However, the nurse must be careful to continue observing the site for any evidence of bleeding (see Family-Centered Care box and Critical Thinking Case Study). After Cardiac Catheterization • Remove pressure dressing the day after catheterization. Cover site with an adhesive bandage strip for several days. • Keep site clean and dry. Avoid tub baths for several days; patient may shower. • Observe site for redness, swelling, drainage, and bleeding. Monitor for fever. Notify health care practitioner if these occur. • Avoid strenuous exercise for several days; patient may attend school. • Resume regular diet without restrictions. • Use acetaminophen or ibuprofen for pain. • Keep follow-up appointments per health care practitioner’s instruction. Adapted from Children’s Hospital (Boston) Cardiovascular Program, 1996. Cardiac Catheterization 1. Evidence—Is there sufficient evidence to draw conclusions about Tommy’s situation? 2. Assumptions—Describe an underlying assumption about each of the following: a. Risks of cardiac catheterization b. Association between vomiting and bleeding after cardiac catheterization 3. What priorities for nursing care should be established for Tommy? The incidence of CHD in children is approximately 5 to 8 per 1000 live births (Park, 2008). About 2 or 3 in 1000 infants will be symptomatic during the first year of life with significant heart disease that requires treatment (Hoffman and Kaplan, 2002). CHD is the major cause of death (other than prematurity) in the first year of life. Although there are more than 35 well-recognized cardiac defects, the most common heart anomaly is ventricular septal defect (VSD). There are typically two classification systems used to categorize congenital heart defects. Traditionally, cyanosis, a physical characteristic, has been used as the distinguishing feature, dividing anomalies into acyanotic defects and cyanotic defects (Fig. 42-2). In clinical practice, this system is problematic because children with acyanotic defects may develop cyanosis. Also, more often, those with cyanotic defects may appear pink and have more clinical signs of HF. A more useful classification system is based on hemodynamic characteristics (blood flow patterns within the heart). These blood flow patterns are (1) increased pulmonary blood flow; (2) decreased pulmonary blood flow; (3) obstruction to blood flow out of the heart; and (4) mixed blood flow, in which saturated and desaturated blood mix within the heart or great arteries. As a comparison, Fig. 42-3 outlines both classification systems. With the hemodynamic classification system, the clinical manifestations of each group are more uniform and predictable. Defects that allow blood flow from the higher-pressure left side of the heart to the lower-pressure right side (left-to-right shunt) result in increased pulmonary blood flow and cause heart failure (HF). Obstructive defects impede blood flow out of the ventricles; whereas obstruction on the left side of the heart results in HF, severe obstruction on the right side causes cyanosis. Defects that cause decreased pulmonary blood flow result in cyanosis. Mixed lesions present a variable clinical picture based on the degree of mixing and amount of pulmonary blood flow; hypoxemia (with or without cyanosis) and HF usually occur together. Using this classification system, the clinical presentation and management of the most common defects are outlined in the following sections and Box 42-1. Box 42-1 Defects with Increased Pulmonary Blood Flow Description—Abnormal opening between the atria, allowing blood from the higher-pressure left atrium to flow into the lower-pressure right atrium. There are three types of ASD: • Ostium primum (ASD 1)—Opening at lower end of septum; may be associated with mitral valve abnormalities • Ostium secundum (ASD 2)—Opening near center of septum • Sinus venosus defect—Opening near junction of superior vena cava and right atrium; may be associated with partial anomalous pulmonary venous connection Pathophysiology—Because left atrial pressure slightly exceeds right atrial pressure, blood flows from the left to the right atrium, causing an increased flow of oxygenated blood into the right side of the heart. Despite the low pressure difference, a high rate of flow can still occur because of low pulmonary vascular resistance and the greater distensibility of the right atrium, which further reduces flow resistance. This volume is well tolerated by the right ventricle because it is delivered under much lower pressure than with a VSD. Although there is right atrial and ventricular enlargement, cardiac failure is unusual in an uncomplicated ASD. Pulmonary vascular changes usually occur only after several decades if the defect is left unrepaired. Clinical manifestations—Patients may be asymptomatic. They may develop HF. There is a characteristic systolic murmur with a fixed split second heart sound. There may also be a diastolic murmur. Patients are at risk for atrial dysrhythmias (probably caused by atrial enlargement and stretching of conduction fibers) and pulmonary vascular obstructive disease and emboli formation later in life from chronically increased pulmonary blood flow. Surgical treatment—Surgical patch closure (pericardial patch or Dacron patch) is done for moderate to large defects. Open repair with cardiopulmonary bypass is usually performed before school age. In addition, the sinus venosus defect requires patch placement, so the anomalous right pulmonary venous return is directed to the left atrium with a baffle. ASD 1 type may require mitral valve repair or, rarely, replacement of the mitral valve. Nonsurgical treatment—ASD 2 closure with a device during cardiac catheterization is becoming commonplace and can be done as an outpatient procedure. The Amplatzer Septal Occluder is most commonly used. Smaller defects that have a rim around them for attachment of the device can be closed with a device; large, irregular defects without a rim require surgical closure. Successful closure in appropriately selected patients yields results similar to those from surgery but involves shorter hospital stays and fewer complications. Patients receive low-dose aspirin for 6 months (Rome and Kreutzer, 2004). Description—Abnormal opening between the right and left ventricles. May be classified according to location: membranous (accounting for 80%); or muscular. May vary in size from a small pinhole to absence of the septum, which results in a common ventricle. VSDs are frequently associated with other defects, such as pulmonary stenosis, transposition of the great vessels, PDA, atrial defects, and COA. Many VSDs (20%-60%) close spontaneously. Spontaneous closure is most likely to occur during the first year of life in children having small or moderate defects. A left-to-right shunt is caused by the flow of blood from the higher-pressure left ventricle to the lower-pressure right ventricle. Pathophysiology—Because of the higher pressure within the left ventricle and because the systemic arterial circulation offers more resistance than the pulmonary circulation, blood flows through the defect into the pulmonary artery. The increased blood volume is pumped into the lungs, which may eventually result in increased pulmonary vascular resistance. Increased pressure in the right ventricle as a result of left-to-right shunting and pulmonary resistance causes the muscle to hypertrophy. If the right ventricle is unable to accommodate the increased workload, the right atrium may also enlarge as it attempts to overcome the resistance offered by incomplete right ventricular emptying. Clinical manifestations—HF is common. There is a characteristic loud holosystolic murmur heard best at the left sternal border. Patients are at risk for BE and pulmonary vascular obstructive disease. • Palliative—Pulmonary artery banding (placement of a band around the main pulmonary artery to decrease pulmonary blood flow) may be done in infants with multiple muscular VSDs or complex anatomy. Improvements in surgical techniques and postoperative care make complete repair in infancy the preferred approach. • Complete repair (procedure of choice)—Small defects are repaired with sutures. Large defects usually require that a knitted Dacron patch be sewn over the opening. CPB is used for both procedures. The approach for the repair is generally through the right atrium and the tricuspid valve. Postoperative complications include residual VSD and conduction disturbances. Nonsurgical treatment—Device closure during cardiac catheterization is being performed in some centers under investigational protocols. One device has been approved for closure of muscular defects, and another is in clinical trials. Early results are encouraging, with successful defect closure and few complications (Rome and Kreutzer, 2004). Prognosis—Risks depend on the location of the defect, the number of defects, and the presence of other associated cardiac defects. Single-membranous defects are associated with low mortality (<2%); multiple muscular defects can carry a higher risk (Jacobs, Mavroudis, Jacobs, et al., 2004). Description—Incomplete fusion of the endocardial cushions. Consists of a low ASD that is continuous with a high VSD and clefts of the mitral and tricuspid valves, which create a large central AV valve that allows blood to flow between all four chambers of the heart. The directions and pathways of flow are determined by pulmonary and systemic resistance, left and right ventricular pressures, and the compliance of each chamber, although flow is generally from left to right. It is the most common cardiac defect in children with Down syndrome. Pathophysiology—The alterations in hemodynamics depend on the severity of the defect and the child’s pulmonary vascular resistance. Immediately after birth, while the newborn’s pulmonary vascular resistance is high, there is minimum shunting of blood through the defect. When this resistance falls, left-to-right shunting occurs and pulmonary blood flow increases. The resultant pulmonary vascular engorgement predisposes the child to development of HF. Clinical manifestations—Patients usually have moderate to severe HF. There is a loud systolic murmur. There may be mild cyanosis that increases with crying. Patients are at high risk for developing pulmonary vascular obstructive disease. • Palliative—Pulmonary artery banding is occasionally done in small infants with severe symptoms. Complete repair in infancy is most common. • Complete repair—Surgical repair consists of patch closure of the septal defects and reconstruction of the AV valve tissue (either repair of the mitral valve cleft or fashioning of two AV valves). Postoperative complications include heart block, HF, mitral regurgitation, dysrhythmias, and pulmonary hypertension. Prognosis—Operative mortality is less than 5% (Jacobs, Mavroudis, Jacobs, et al., 2004). A potential later problem is mitral regurgitation, which may require valve replacement. Description—Failure of the fetal ductus arteriosus (artery connecting the aorta and pulmonary artery) to close within the first weeks of life. The continued patency of this vessel allows blood to flow from the higher-pressure aorta to the lower-pressure pulmonary artery, which causes a left-to-right shunt. Pathophysiology—The hemodynamic consequences of PDA depend on the size of the ductus and the pulmonary vascular resistance. At birth, the resistance in the pulmonary and systemic circulations is almost identical, so that the resistance in the aorta and pulmonary artery is equalized. As the systemic pressure comes to exceed the pulmonary pressure, blood begins to shunt from the aorta across the duct to the pulmonary artery (left-to-right shunt). The additional blood is recirculated through the lungs and returned to the left atrium and left ventricle. The effects of this altered circulation are increased workload on the left side of the heart, increased pulmonary vascular congestion and possibly resistance, and potentially increased right ventricular pressure and hypertrophy. Clinical manifestations—Patients may be asymptomatic or show signs of HF. There is a characteristic machinery-like murmur. A widened pulse pressure and bounding pulses result from runoff of blood from the aorta to the pulmonary artery. Patients are at risk for BE and pulmonary vascular obstructive disease in later life from chronic excessive pulmonary blood flow. Medical management—Administration of indomethacin (a prostaglandin inhibitor) has proved successful in closing a PDA in preterm infants and some newborns. Surgical treatment—Surgical division or ligation of the patent vessel is performed via a left thoracotomy. In a newer technique, video-assisted thoracoscopic surgery, a thoracoscope and instruments are inserted through three small incisions on the left side of the chest to place a clip on the ductus. The technique is used in some centers and eliminates the need for a thoracotomy, thereby speeding postoperative recovery. Nonsurgical treatment—Coils to occlude the PDA are placed in the catheterization laboratory in many centers. Preterm or small infants (with small-diameter femoral arteries) and patients with large or unusual PDAs may require surgery. Prognosis—Both surgical and nonsurgical procedures can be done at low risk with less than 1% mortality. PDA closure in very preterm infants has a higher mortality rate because of the additional significant medical problems. In this group of cardiac defects, intracardiac communications along the septum or an abnormal connection between the great arteries allows blood to flow from the higher pressure left side of the heart to the lower pressure right side of the heart. Increased blood volume on the right side of the heart increases pulmonary blood flow at the expense of systemic blood flow. Clinically, patients demonstrate signs and symptoms of HF. ASD, VSD, and patent ductus arteriosus are typical anomalies in this group (see Box 42-1). Obstructive defects are those in which blood exiting the heart meets an area of anatomic narrowing (stenosis), causing obstruction to blood flow. The pressure in the ventricle and in the great artery before the obstruction is increased, and the pressure in the area beyond the obstruction is decreased. The location of the narrowing is usually near the valve (Fig. 42-4), as follows: • Valvular—At the site of the valve itself • Subvalvular—Narrowing in the ventricle below the valve (also referred to as the ventricular outflow tract) • Supravalvular—Narrowing in the great artery above the valve Coarctation of the aorta (narrowing of the aortic arch), aortic stenosis, and pulmonic stenosis are typical defects in this group (Box 42-2). Hemodynamically, there is a pressure load on the ventricle and decreased cardiac output. Clinically, infants and children exhibit signs of HF. Children with mild obstruction may be asymptomatic. Rarely, as in severe pulmonic stenosis, hypoxemia may be seen. Box 42-2 Obstructive Defects Description—Localized narrowing near the insertion of the ductus arteriosus, which results in increased pressure proximal to the defect (head and upper extremities) and decreased pressure distal to the obstruction (body and lower extremities). Pathophysiology—The effect of a narrowing within the aorta is increased pressure proximal to the defect (upper extremities) and decreased pressure distal to it (lower extremities). Clinical manifestations—The patient may have high blood pressure and bounding pulses in the arms, weak or absent femoral pulses, and cool lower extremities with lower blood pressure. There are signs of HF in infants. In infants with critical coarctation, the hemodynamic condition may deteriorate rapidly with severe acidosis and hypotension. Mechanical ventilation and inotropic support are often necessary before surgery. Older children may experience dizziness, headaches, fainting, and epistaxis resulting from hypertension. Patients are at risk for hypertension, ruptured aorta, aortic aneurysm, and stroke. Surgical treatment—Surgical repair is the treatment of choice for infants younger than 6 months and for patients with long-segment stenosis or complex anatomy; it may be performed for all patients with coarctation. Repair is by resection of the coarctated portion with an end-to-end anastomosis of the aorta or enlargement of the constricted section using a graft of prosthetic material or a portion of the left subclavian artery. Because this defect is outside the heart and pericardium, cardiopulmonary bypass is not required and a thoracotomy incision is used. Postoperative hypertension is treated with intravenous sodium nitroprusside, esmolol, or milrinone followed by oral medications, such as ACE inhibitors or beta blockers. Residual permanent hypertension after repair of COA seems to be related to age and time of repair. To prevent both hypertension at rest and exercise-provoked systemic hypertension after repair, elective surgery for COA is advised within the first 2 years of life. There is a 15% to 30% risk for recurrence in patients who underwent surgical repair as infants (Beekman, 2001). Percutaneous balloon angioplasty techniques have proved to be effective in relieving residual postoperative coarctation gradients. Nonsurgical treatment—Balloon angioplasty is being performed as a primary intervention for COA in older infants and children. In adolescents, stents may be placed in the aorta to maintain patency. Recent studies have demonstrated that balloon angioplasty is effective in children and that aneurysm formation is rare. The high restenosis rate in young infants limits its application in this group (Rome and Kreutzer, 2004). Prognosis—Mortality is less than 5% in patients with isolated coarctation; the risk is increased in infants with other complex cardiac defects (Jacobs, Mavroudis, Jacobs, et al., 2004). Description—Narrowing or stricture of the aortic valve, causing resistance to blood flow in the left ventricle, decreased cardiac output, left ventricular hypertrophy, and pulmonary vascular congestion. The prominent anatomic consequence of AS is the hypertrophy of the left ventricular wall, which eventually leads to increased end-diastolic pressure, resulting in pulmonary venous and pulmonary arterial hypertension. Left ventricular hypertrophy also interferes with coronary artery perfusion and may result in myocardial infarction or scarring of the papillary muscles of the left ventricle, which causes mitral insufficiency. Valvular stenosis, the most common type, is usually caused by malformed cusps that result in a bicuspid rather than tricuspid valve or fusion of the cusps. Subvalvular stenosis is a stricture caused by a fibrous ring below a normal valve; supravalvular stenosis occurs infrequently. Valvular AS is a serious defect for the following reasons: (1) the obstruction tends to be progressive; (2) sudden episodes of myocardial ischemia, or low cardiac output, can result in sudden death; and (3) surgical repair rarely results in a normal valve. This is one of the rare instances in which strenuous physical activity may be curtailed because of the cardiac condition. Pathophysiology—A stricture in the aortic outflow tract causes resistance to ejection of blood from the left ventricle. The extra workload on the left ventricle causes hypertrophy. If left ventricular failure develops, left atrial pressure will increase; this causes increased pressure in the pulmonary veins, which results in pulmonary vascular congestion (pulmonary edema). Clinical manifestations—Newborns with critical AS demonstrate signs of decreased cardiac output with faint pulses, hypotension, tachycardia, and poor feeding. Children show signs of exercise intolerance, chest pain, and dizziness when standing for a long period. A systolic ejection murmur may or may not be present. Patients are at risk for BE, coronary insufficiency, and ventricular dysfunction. Surgical treatment—Aortic valvotomy is performed under inflow occlusion. Used rarely because balloon dilation in the catheterization laboratory is the first-line procedure. Newborns with critical AS and small left-sided structures may undergo a stage 1 Norwood procedure (see Hypoplastic Left Heart Syndrome, Box 42-4). Prognosis—Aortic valve replacement offers a good treatment option and may lead to normalization of left ventricular size and function (Arnold, Ley-Zaporozhan, Ley, et al., 2008). Results of aortic valvotomy in older children are very good, with mortality and morbidity close to 0% (Shanmugam, MacArthur, and Pollock, 2005). However, aortic valvotomy remains a palliative procedure and approximately 25% of patients require additional surgery within 10 years for recurrent stenosis. A valve replacement may be required at the second procedure. An aortic homograft with a valve may also be used (extended aortic root replacement), or the pulmonary valve may be moved to the aortic position and replaced with a homograft valve (Ross procedure). Nonsurgical treatment—The narrowed valve is dilated using balloon angioplasty in the catheterization laboratory. This procedure is usually the first intervention. Prognosis—Complications include aortic insufficiency or valvular regurgitation, tearing of the valve leaflets, and loss of pulse in the catheterized limb. Surgical treatment—Procedure may involve incising a membrane if one exists or cutting the fibromuscular ring. If the obstruction results from narrowing of the left ventricular outflow tract and a small aortic valve annulus, a patch may be required to enlarge the entire left ventricular outflow tract and annulus and replace the aortic valve; this is known as the Konno procedure. Prognosis—Mortality from surgical repairs of subvalvular AS is less than 5% in major centers; however, about 20% of these patients develop recurrent subaortic stenosis and require additional surgery (Freed, 2001). Description—Narrowing at the entrance to the pulmonary artery. Resistance to blood flow causes right ventricular hypertrophy and decreased pulmonary blood flow. Pulmonary atresia is the extreme form of PS in that there is total fusion of the commissures and no blood flows to the lungs. The right ventricle may be hypoplastic. Pathophysiology—When PS is present, resistance to blood flow causes right ventricular hypertrophy. If right ventricular failure develops, right atrial pressure will increase and this may result in reopening of the foramen ovale, shunting of unoxygenated blood into the left atrium, and systemic cyanosis. If PS is severe, HF occurs and systemic venous engorgement will be noted. An associated defect such as a PDA partially compensates for the obstruction by shunting blood from the aorta to the pulmonary artery and into the lungs. Clinical manifestations—Patients may be asymptomatic; some have mild cyanosis or HF. Progressive narrowing causes increased symptoms. Newborns with severe narrowing are cyanotic. A loud systolic ejection murmur at the upper left sternal border may be present. However, in severely ill patients, the murmur may be much softer because of decreased cardiac output and shunting of blood. Cardiomegaly is evident on chest radiography. Patients are at risk for BE. Surgical treatment—In infants, transventricular (closed) valvotomy (Brock procedure). In children, pulmonary valvotomy with CPB. Need for surgical treatment is rare with widespread use of balloon angioplasty techniques. Nonsurgical treatment—Balloon angioplasty in the cardiac catheterization laboratory to dilate the valve. A catheter is inserted across the stenotic pulmonic valve into the pulmonary artery, and a balloon at the end of the catheter is inflated and rapidly passed through the narrowed opening (see figure at right). The procedure is associated with few complications and has proved to be highly effective. It is the treatment of choice for discrete PS in most centers and can be done safely in neonates. Prognosis—The risk is low for both surgical and nonsurgical procedures; mortality is lower than 1% and slightly higher in neonates (Latson, 2001). Both balloon dilation and surgical valvotomy leave the pulmonic valve incompetent because they involve opening the fused valve leaflets; however, these patients are clinically asymptomatic. Long-term problems with restenosis or valve incompetence may occur. In this group of defects, there is obstruction of pulmonary blood flow and an anatomic defect (ASD or VSD) between the right and left sides of the heart (Fig. 42-5). Because blood has difficulty exiting the right side of the heart via the pulmonary artery, pressure on the right side increases, exceeding left-sided pressure. This allows desaturated blood to shunt right to left, causing desaturation in the left side of the heart and in the systemic circulation. Clinically, these patients have hypoxemia and usually appear cyanotic. Tetralogy of Fallot and tricuspid atresia are the most common defects in this group (Box 42-3). Box 42-3 Defects with Decreased Pulmonary Blood Flow Description—The classic form includes four defects: (1) VSD, (2) PS, (3) overriding aorta, and (4) right ventricular hypertrophy. Pathophysiology—The alteration in hemodynamics varies widely, depending primarily on the degree of PS but also on the size of the VSD and the pulmonary and systemic resistance to flow. Because the VSD is usually large, pressures may be equal in the right and left ventricles. Therefore the shunt direction depends on the difference between pulmonary and systemic vascular resistance. If pulmonary vascular resistance is higher than systemic resistance, the shunt is from right to left. If systemic resistance is higher than pulmonary resistance, the shunt is from left to right. PS decreases blood flow to the lungs and consequently the amount of oxygenated blood that returns to the left side of the heart. Depending on the position of the aorta, blood from both ventricles may be distributed systemically. Clinical manifestations—Some infants may be acutely cyanotic at birth; others have mild cyanosis that progresses over the first year of life as the PS worsens. There is a characteristic systolic murmur that is often moderate in intensity. There may be acute episodes of cyanosis and hypoxia, called blue spells or tet spells (see p. 1337). Anoxic spells occur when the infant’s oxygen requirements exceed the blood supply, usually during crying or after feeding. Patients are at risk for emboli, seizures, and loss of consciousness or sudden death after an anoxic spell. • Palliative shunt—In infants who cannot undergo primary repair, a palliative procedure to increase pulmonary blood flow and increase oxygen saturation may be performed. The preferred procedure is a modified Blalock-Taussig shunt operation, which provides blood flow to the pulmonary arteries from the left or right subclavian artery via a tube graft (see Table 42-2). In general, however, shunts are avoided because they may result in pulmonary artery distortion. • Complete repair—Elective repair is usually performed in the first year of life. Indications for repair include increasing cyanosis and the development of hypercyanotic spells. Complete repair involves closure of the VSD and resection of the infundibular stenosis, with placement of a pericardial patch to enlarge the RVOT. In some repairs, the patch may extend across the pulmonary valve annulus (transannular patch), making the pulmonary valve incompetent. The procedure requires a median sternotomy and the use of cardiopulmonary bypass. Prognosis—The operative mortality for total correction of tetralogy of Fallot is less than 3% (Jacobs, Mavroudis, Jacobs, et al., 2004). With improved surgical techniques, there is a lower incidence of dysrhythmias and sudden death; surgical heart block is rare. Heart failure may occur postoperatively. Description—The tricuspid valve fails to develop; consequently there is no communication from the right atrium to the right ventricle. Blood flows through an ASD or a patent foramen ovale to the left side of the heart and through a VSD to the right ventricle and out to the lungs. The condition is often associated with PS and TGA. There is complete mixing of unoxygenated and oxygenated blood in the left side of the heart, which results in systemic desaturation, and varying amounts of pulmonary obstruction, which causes decreased pulmonary blood flow. Pathophysiology—At birth, the presence of a patent foramen ovale (or other atrial septal opening) is required to permit blood flow across the septum into the left atrium; the PDA allows blood flow to the pulmonary artery into the lungs for oxygenation. A VSD allows a modest amount of blood to enter the right ventricle and pulmonary artery for oxygenation. Pulmonary blood flow usually is diminished. Clinical manifestations—Cyanosis is usually seen in the newborn period. There may be tachycardia and dyspnea. Older children have signs of chronic hypoxemia with clubbing. Therapeutic management—For neonates whose pulmonary blood flow depends on the patency of the ductus arteriosus, a continuous infusion of prostaglandin E1 is started at 0.1 mcg/kg/min until surgical intervention can be arranged. Surgical treatment—Palliative treatment is the placement of a shunt (pulmonary-to-systemic artery anastomosis) to increase blood flow to the lungs. If the ASD is small, an atrial septostomy is performed during cardiac catheterization. Some children have increased pulmonary blood flow and require pulmonary artery banding to lessen the volume of blood to the lungs. A bidirectional Glenn shunt (cavopulmonary anastomosis) may be performed at 4 to 9 months as a second stage. • Modified Fontan procedure—Systemic venous return is directed to the lungs without a ventricular pump through surgical connections between the right atrium and the pulmonary artery. A fenestration (opening) is sometimes made in the right atrial baffle to relieve pressure. The patient must have normal ventricular function and a low pulmonary vascular resistance for the procedure to be successful. The modified Fontan procedure separates oxygenated and unoxygenated blood inside the heart and eliminates the excess volume load on the ventricle but does not restore normal anatomy or hemodynamics. This operation is also the final stage in the correction of many complex defects with a functional single ventricle, including hypoplastic left heart syndrome.
Cardiovascular Dysfunction
Cardiovascular Dysfunction
History and Physical Examination
Inspection
Diagnostic Evaluation
Cardiac Catheterization
Care Management
Preprocedural Care
Postprocedural Care
 Family-Centered Care
Family-Centered Care
 Critical Thinking Case Study
Critical Thinking Case Study
Congenital Heart Disease
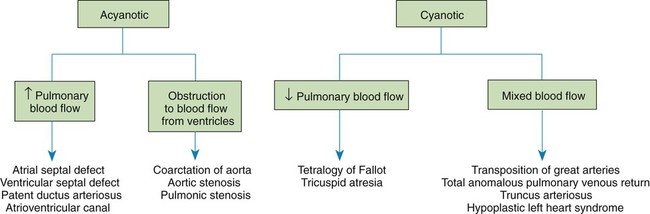
Classification of Defects
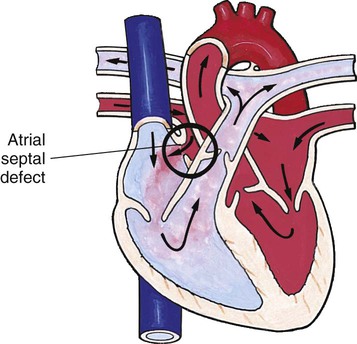
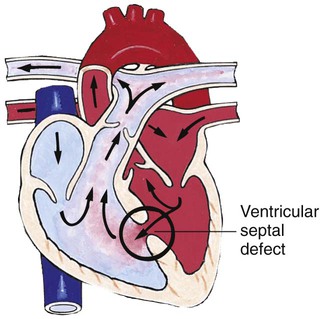
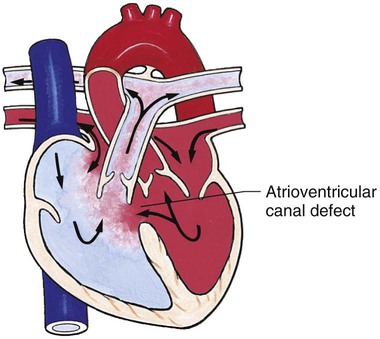
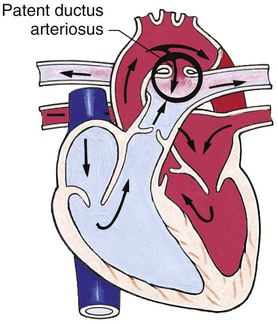
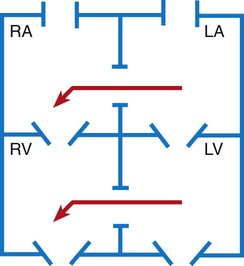
Defects with Increased Pulmonary Blood Flow
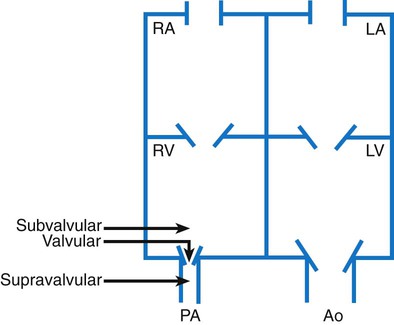
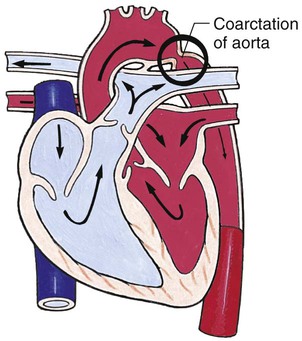
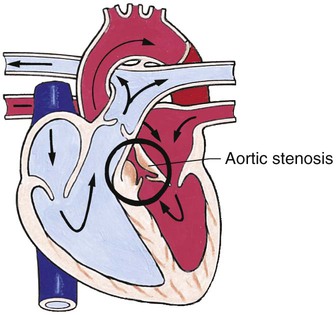
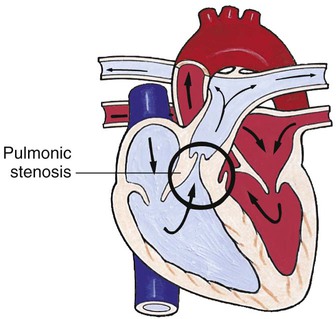
Obstructive Defects
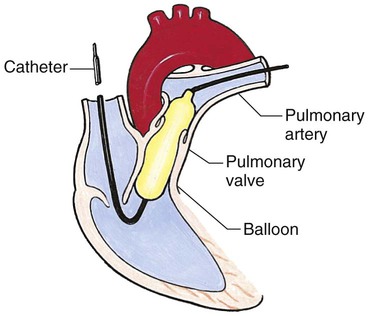
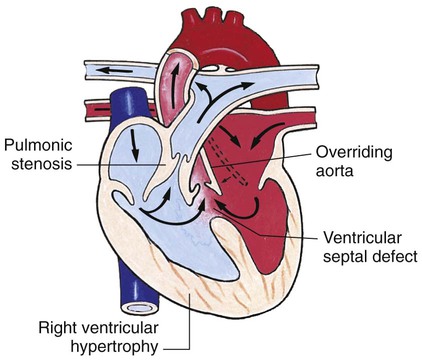
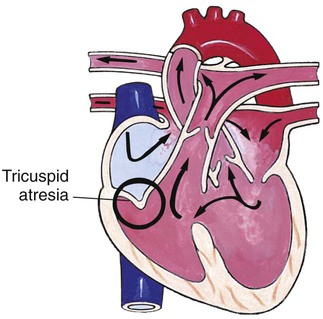
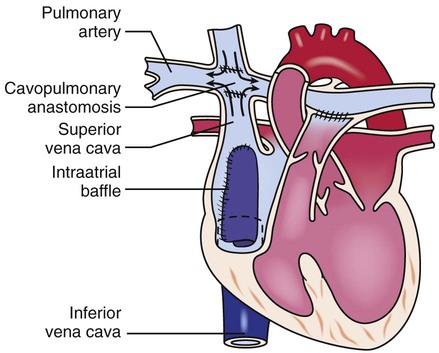
Defects with Decreased Pulmonary Blood Flow
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Cardiovascular Dysfunction
 Nursing Alert
Nursing Alert Nursing Alert
Nursing Alert
Get Clinical Tree app for offline access