Discuss cellular physiology in relation to drug therapy.
 Describe the main pathways and mechanisms by which drugs cross biologic membranes and move through the body.
Describe the main pathways and mechanisms by which drugs cross biologic membranes and move through the body.
 Explain each process of pharmacokinetics.
Explain each process of pharmacokinetics.
 Discuss the clinical usefulness of measuring serum drug levels.
Discuss the clinical usefulness of measuring serum drug levels.
 Describe major characteristics of the receptor theory of drug action.
Describe major characteristics of the receptor theory of drug action.
 Differentiate between agonist drugs and antagonist drugs.
Differentiate between agonist drugs and antagonist drugs.
 List drug-related and patient-related variables that affect drug actions.
List drug-related and patient-related variables that affect drug actions.
 Discuss mechanisms and potential effects of drug–drug interactions.
Discuss mechanisms and potential effects of drug–drug interactions.
 Identify signs and symptoms that may occur with adverse drug effects on major body systems.
Identify signs and symptoms that may occur with adverse drug effects on major body systems.
 Discuss general management of drug overdose and toxicity.
Discuss general management of drug overdose and toxicity.
 Discuss selected drug antidotes.
Discuss selected drug antidotes.
Clinical Application Case Study
Doris Green, an 89-year-old widow with cardiovascular and renal disease, takes a number of medications, including three medications to control her hypertension. She has recently switched to a new antihypertensive medication. She prides herself on being independent and able to manage on her own, despite failing memory and failing health.
You, as a home health nurse, visit Mrs. Green to assess the therapeutic and adverse effects of her antihypertensive medications and her adherence to the prescribed medical regimen. You plan to measure her blood pressure to check that it is within normal limits (for Mrs. Green), as well as assess her understanding of her antihypertensive medications and dosing regimen.
KEY TERMS
Absorption: process that occurs from the time a drug enters the body to the time it enters the bloodstream to be circulated
Agonist: drug that produces effects similar to those produced by naturally occurring hormones, neurotransmitters, and other substances
Antagonist: drug that inhibits cell function by occupying receptor sites
Antidote: substance that relieves, prevents, or counteracts the effect of a poison
Bioavailability: portion of a drug dose that reaches the systemic circulation and is available to act on body cells
Biotransformation: when drugs are altered from their original form into a new form by the body; also referred to as metabolism
Distribution: transport of drug molecules within the body; after a drug is injected or absorbed into the bloodstream, it is carried by the blood and tissue fluids to its sites of action, metabolism, and excretion
Enterohepatic recirculation: drugs or metabolites that are excreted in bile, reabsorbed from the small intestine, returned to the liver, metabolized, and eventually excreted in urine.
Enzyme induction: production of larger amounts of drug-metabolizing enzymes by liver cells; process accelerates drug metabolism because larger amounts of the enzymes (and more binding sites) allow larger amounts of a drug to be metabolized during a given time
Enzyme inhibition: process in which a molecule binds to enzymes and inhibits their activity
Excretion: elimination of a drug from the body; effective excretion requires adequate functioning of the circulatory system and of the organs of excretion (kidneys, bowel, lungs, and skin)
First-pass effect: initial metabolism of some oral drugs as they are carried from the intestine to the liver by the portal circulatory system prior to reaching the systemic circulation for distribution to site of action
Hypersensitivity: immune-mediated reaction to a drug
Loading dose: dose larger than the regular prescribed daily dosage of a medication; used to attain a therapeutic blood level
Maintenance dose: quantity of drug that is needed to keep blood levels and/or tissue levels at a steady state or constant level
Nephrotoxicity: toxic or damaging effect of a substance on the kidney; potentially serious because renal damage interferes with drug excretion, causing drug accumulation and increased adverse effects
Pharmacodynamics: reactions between living systems and drugs; drug actions on target cells and the resulting alterations in cellular biochemical reactions and functions
Pharmacokinetics: drug movement through the body to reach sites of action, metabolism, and excretion
Prodrugs: initially inactive drugs that exert no pharmacologic effects until they are metabolized
Serum drug level: laboratory measurement of the amount of a drug in the blood at a particular time
Serum half-life: time required for the serum concentration of a drug to decrease by 50%; also called elimination half-life
Introduction
All body functions, disease processes, and most drug actions occur at the cellular level. Drugs are chemicals that alter basic processes in body cells. They can stimulate or inhibit normal cellular functions; however, they cannot change the type of function that occurs normally. To act on body cells, drugs given for systemic effects must reach adequate concentrations in the blood and other tissue fluids surrounding the cells. Thus, they must enter the body and be circulated to their sites of action (target cells). After they act on cells, they must be eliminated from the body.
How do systemic drugs reach, interact with, and leave body cells? How do people respond to drugs? The answers to these questions are derived from cellular physiology, pathways and mechanisms of drug transport, pharmacokinetics, pharmacodynamics, and other basic concepts and processes that form the foundation of rational drug therapy and the content of this chapter.
Cellular Physiology
Cells are dynamic, busy “factories” (Box 2.1, Fig. 2.1) that take in raw materials, manufacture products required to maintain bodily functions, and deliver those products to their appropriate destinations in the body. Although cells differ from one tissue to another, their common characteristics include the ability to
• Exchange materials with their immediate environment
• Obtain energy from nutrients
• Synthesize hormones, neurotransmitters, enzymes, structural proteins, and other complex molecules
• Reproduce
• Communicate with one another via various biologic chemicals, such as neurotransmitters and hormones
BOX 2.1 Cell Structures and Functions
Protoplasm, which constitutes the internal environment of body cells, is composed of water, electrolytes (potassium, magnesium, phosphate, sulfate, bicarbonate), proteins, lipids, and carbohydrates. Water makes up 70% to 85% of most cells; cellular enzymes, electrolytes, and other chemicals are dissolved or suspended in the water. Electrolytes provide chemicals for cellular reactions and are required for some processes (e.g., transmission of electrochemical impulses in nerve and muscle cells). Proteins consist of “physical” proteins that form the structure of cells and “chemical” proteins that function mainly as enzymes within the cell. Lipids, mainly phospholipids and cholesterol, form the membranes that separate structures inside the cell and the cell itself from surrounding cells and body fluids. Carbohydrates play a major role in cell nutrition. Glucose is present in extracellular fluid and is readily available to supply the cell’s need for energy. In addition, a small amount of carbohydrate is stored within the cell as glycogen, which can be rapidly converted back to glucose when needed.
The nucleus regulates the types and amounts of proteins, enzymes, and other substances to be produced. The cytoplasm surrounds the nucleus and contains the working units of the cell. The cytosol, the clear fluid portion of the cytoplasm, contains dissolved proteins, electrolytes, and glucose. The endoplasmic reticulum (ER) contains ribosomes, which synthesize proteins, including enzymes that synthesize glycogen, triglycerides, and steroids and those that metabolize drugs and other chemicals. The ER is also important in the production of hormones by glandular cells and the production of plasma proteins by liver cells. Most proteins produced by the ribosomes are released into the cytosol where they act as enzymes or structural proteins of the cell. The Golgi complex stores the substances produced by the ER. It also packages these substances into secretory granules, which then move out of the Golgi complex into the cytoplasm and, after a stimulus, are released from the cell. Mitochondria generate energy for cellular activities and require oxygen. Lysosomes are membrane-enclosed vesicles that contain enzymes capable of digesting nutrients (proteins, carbohydrates, fats), damaged cellular structures, foreign substances (e.g., bacteria), and the cell itself. When a cell becomes worn out or damaged, the membrane around the lysosome breaks and the enzymes (hydrolases) are released. However, lysosomal contents also are released into extracellular spaces and may destroy surrounding cells. Normally, the enzymes are inactivated by enzyme inhibitors, and excessive tissue destruction is prevented.
The cell membrane, a complex structure of phospholipids, proteins, cholesterol, and carbohydrates, separates intracellular contents from the extracellular environment; provides receptors for hormones and other biologically active substances; participates in electrical events that occur in nerve and muscle cells; and helps regulate growth and proliferation.
The cell membrane covers the entire surface of the cell and consists of a thin, double layer of lipids interspersed with proteins. The lipid layer is composed of phospholipid (fatty acid and phosphate) molecules. The proteins are usually combined with a carbohydrate and called glycoproteins. Some proteins provide structural pores through which water and water-soluble substances (e.g., sodium, potassium, and calcium ions) can diffuse between extracellular and intracellular fluids. Other proteins act as carriers to transport substances through the cell membrane. Still others act to regulate intracellular function or as enzymes to catalyze chemical reactions within the cell.
Carbohydrates in the cell membrane occur mainly in combination with proteins (glycoproteins) or lipids (glycolipids). Glycoproteins (composed of carbohydrate around a small, inner core of protein and called proteoglycans) often are attached to and cover the entire outside surface of the cell. As a result, the carbohydrate molecules are free to interact with extracellular substances and perform several important functions. First, many have a negative electrical charge that repels other negatively charged substances. Second, the “carbohydrate coat” of some cells attaches to the carbohydrate coat of other cells and thereby connects cells to each other. Third, many of the carbohydrates act as receptor molecules for binding hormones (e.g., insulin). The receptor-hormone combination then activates the attached inner core of protein to perform its enzymatic or other functions in the cell.
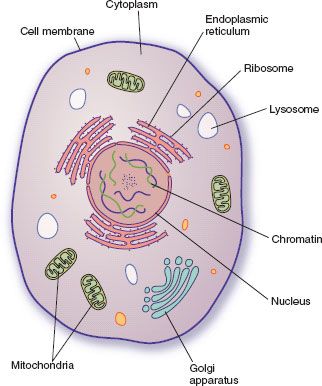
Figure 2.1 Cytoplasmic organelles of the cell.
Drug Transport Through Cell Membranes
Drugs must reach and interact with or cross the cell membrane to stimulate or inhibit cellular function. Most drugs are given to affect body cells that are distant from the sites of administration (i.e., systemic effects). To move through the body and reach their sites of action, metabolism, and excretion (Fig. 2.2), drug molecules must cross numerous cell membranes. For example, molecules of most oral drugs must cross the membranes of cells in the gastrointestinal (GI) tract, liver, and capillaries to reach the bloodstream, circulate to their target cells, leave the bloodstream and attach to receptors on cells, perform their action, return to the bloodstream, circulate to the liver, reach drug-metabolizing enzymes in liver cells, reenter the bloodstream (usually as metabolites), circulate to the kidneys, and be excreted in urine. Box 2.2 and Figure 2.3 describe the transport pathways and mechanisms used to move drug molecules through the body.
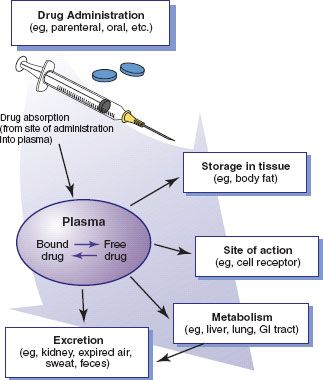
Figure 2.2 Entry and movement of drug molecules through the body to sites of action, metabolism, and excretion.
BOX 2.2 Drug Transport Pathways and Mechanisms
Pathways
There are three main pathways of drug movement across cell membranes. The most common pathway is direct penetration of the membrane by lipid-soluble drugs, which are able to dissolve in the lipid layer of the cell membrane. Most systemic drugs are formulated to be lipid soluble so they can move through cell membranes, even oral tablets and capsules that must be sufficiently water soluble to dissolve in the aqueous fluids of the stomach and small intestine.
A second pathway involves passage through protein channels that go all the way through the cell membrane. Only a few drugs are able to use this pathway because most drug molecules are too large to pass through the small channels. Small ions (e.g., sodium and potassium) use this pathway, but their movement is regulated by specific channels with a gating mechanism (a flap of protein that opens briefly to allow ion movement and then closes).
The third pathway involves carrier proteins that transport molecules from one side of the cell membrane to the other. All of the carrier proteins are selective in the substances they transport; a drug’s chemical structure determines which carrier will transport it.
Mechanisms
Once absorbed into the body, drugs are transported to and from target cells by passive diffusion, facilitated diffusion, and active transport.
Passive diffusion, the most common mechanism, involves movement of a drug from an area of higher concentration to one of lower concentration. For example, after oral administration, the initial concentration of a drug is higher in the gastrointestinal tract than in the blood. This promotes movement of the drug into the bloodstream. When the drug is circulated, the concentration is higher in the blood than in body cells, so that the drug moves (from capillaries) into the fluids surrounding the cells or into the cells themselves. Passive diffusion continues until a state of equilibrium is reached between the amount of drug in the tissues and the amount in the blood.
Facilitated diffusion is a similar process, except that drug molecules combine with a carrier substance, such as an enzyme or other protein.
In active transport, drug molecules are moved from an area of lower concentration to one of higher concentration. This process requires a carrier substance and the release of cellular energy.
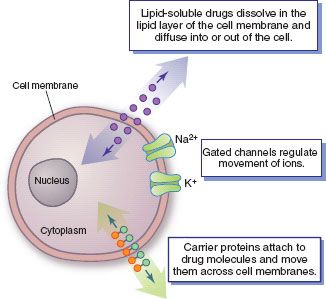
Figure 2.3 Drug transport pathways. Drug molecules cross cell membranes to move into and out of body cells by directly penetrating the lipid layer, diff using through open or gated channels, or attaching to carrier proteins.
Pharmacokinetics
Pharmacokinetics involves drug movement through the body (i.e., “what the body does to the drug”) to reach sites of action, metabolism, and excretion. Specific processes are absorption, distribution, metabolism, and excretion. Metabolism and excretion are often grouped together as drug elimination or clearance mechanisms. Overall, these processes largely determine serum drug levels; onset, peak, and duration of drug actions; therapeutic and adverse effects; and other important aspects of drug therapy.
Absorption
Absorption is the process that occurs from the time a drug enters the body to the time it enters the bloodstream to be circulated. Onset of drug action is largely determined by the rate of absorption; intensity is determined by the extent of absorption. Numerous factors affect the rate and extent of drug absorption, including dosage form, route of administration, blood flow to the site of administration, GI function, the presence of food or other drugs, and other variables. Dosage form is a major determinant of a drug’s bioavailability (the portion of a dose that reaches the systemic circulation and is available to act on body cells). An intravenous (IV) drug is virtually 100% bioavailable. In contrast, an oral drug is virtually always less than 100% bioavailable because some of it is not absorbed from the GI tract and some goes to the liver and is partially metabolized before reaching the systemic circulation.
Most oral drugs must be swallowed, dissolved in gastric fluid, and delivered to the small intestine (which has a large surface area for absorption of nutrients and drugs) before they are absorbed. Liquid medications are absorbed faster than tablets or capsules because they need not be dissolved. Rapid movement through the stomach and small intestine may increase drug absorption by promoting contact with absorptive mucous membrane; it also may decrease absorption because some drugs may move through the small intestine too rapidly to be absorbed. For many drugs, the presence of food in the stomach slows the rate of absorption and may decrease the amount of drug absorbed.
Drugs injected into subcutaneous (subcut) or intramuscular (IM) tissues are usually absorbed more rapidly than oral drugs because they move directly from the injection site to the bloodstream. Absorption is rapid from IM sites because muscle tissue has an abundant blood supply. Drugs injected intravenously do not need to be absorbed because they are placed directly into the bloodstream.
Other absorptive sites include the skin, mucous membranes, and lungs. Most drugs applied to the skin are given for local effects (e.g., sunscreens). Systemic absorption is minimal from intact skin but may be considerable when the skin is inflamed or damaged. Also, some drugs are formulated in adhesive skin patches for absorption through the skin (e.g., clonidine, fentanyl, nitroglycerin). Some drugs applied to mucous membranes also are given for local effects. However, systemic absorption occurs from the mucosa of the oral cavity, nose, eye, vagina, and rectum. Drugs absorbed through mucous membranes pass directly into the bloodstream. The lungs have a large surface area for absorption of anesthetic gases and a few other drugs.
Distribution
Distribution involves the transport of drug molecules within the body. After a drug is injected or absorbed into the bloodstream, it is carried by the blood and tissue fluids to its sites of action, metabolism, and excretion. Most drug molecules enter and leave the bloodstream at the capillary level, through gaps between the cells that form capillary walls. Distribution depends largely on the adequacy of blood circulation. Drugs are distributed rapidly to organs receiving a large blood supply, such as the heart, liver, and kidneys. Distribution to other internal organs, muscle, fat, and skin is usually slower.
Protein binding is an important factor in drug distribution (Fig. 2.4). Most drugs form a compound with plasma proteins, mainly albumin, which act as carriers. Drug molecules bound to plasma proteins are pharmacologically inactive because the large size of the complex prevents their leaving the bloodstream through the small openings in capillary walls and reaching their sites of action, metabolism, and excretion. Only the free or unbound portion of a drug acts on body cells. As the free drug acts on cells, the decrease in plasma drug levels causes some of the bound drug to be released.
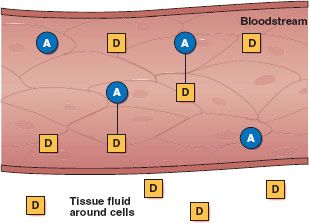
Figure 2.4 Plasma proteins, mainly albumin (A), act as carriers for drug molecules (D). Bound drug (A–D) stays in the bloodstream and is pharmacologically inactive. Free drug (D) can leave the bloodstream and act on body cells.
Protein binding allows part of a drug dose to be stored and released as needed. Some drugs also are stored in muscle, fat, or other body tissues and released gradually when plasma drug levels fall. These storage mechanisms maintain lower, more consistent blood levels and reduce the risk of toxicity. Drugs that are highly bound to plasma proteins or stored extensively in other tissues have a long duration of action.
Drug distribution into the central nervous system (CNS) is limited because the blood–brain barrier, which is composed of capillaries with tight walls, limits movement of drug molecules into brain tissue. This barrier usually acts as a selectively permeable membrane to protect the CNS. However, it also can make drug therapy for CNS disorders more difficult because drugs must pass through cells of the capillary wall rather than between cells. As a result, only drugs that are lipid soluble or have a transport system can cross the blood–brain barrier and reach therapeutic concentrations in brain tissue.
Drug distribution during pregnancy and lactation is also an important consideration (see Chap. 6). During pregnancy, most drugs cross the placenta and may affect the fetus. During lactation, many drugs enter breast milk and may affect the nursing infant.
Metabolism
Metabolism, or biotransformation, is the method by which drugs are inactivated or biotransformed by the body. Most often, an active drug is changed into inactive metabolites, which are then excreted. Some active drugs yield metabolites that are also active and that continue to exert their effects on body cells until they are metabolized further or excreted. Other drugs (called prodrugs) are initially inactive and exert no pharmacologic effects until they are metabolized. Most drugs are lipid soluble, a characteristic that aids their movement across cell membranes. However, the kidneys can excrete only water-soluble substances. Therefore, one function of metabolism is to convert fat-soluble drugs into water-soluble metabolites. Hepatic drug metabolism or clearance is a major mechanism for terminating drug action and eliminating drug molecules from the body.
Most drugs are metabolized by cytochrome P450 (CYP) enzymes in the liver. Red blood cells, plasma, kidneys, lungs, and GI mucosa also contain drug-metabolizing enzymes. The CYP system consists of several groups of enzymes, some of which metabolize endogenous substances and some of which metabolize drugs. The drug-metabolizing groups are labeled CYP1, CYP2, and CYP3. Individual members of the groups usually metabolize specific drugs; more than one enzyme participates in the metabolism of some drugs. In terms of importance in drug metabolism, the CYP3A4 enzymes are thought to metabolize about 50% of drugs; CYP2D6 enzymes about 25%; CYP2C8/9 about 15%; and CYP1A2, 2C19, 2A6, and 2E1 in decreasing order for the remaining 10%.
CYP enzymes are complex proteins with binding sites for drug molecules (and endogenous substances). They catalyze the chemical reactions of oxidation, reduction, hydrolysis, and conjugation with endogenous substances, such as glucuronic acid or sulfate. With chronic administration, some drugs stimulate liver cells to produce larger amounts of drug-metabolizing enzymes. This enzyme induction accelerates drug metabolism because larger amounts of the enzymes (and more binding sites) allow larger amounts of a drug to be metabolized during a given period. As a result, larger doses of the rapidly metabolized drug may be required to produce or maintain therapeutic effects. Rapid metabolism may also increase the production of toxic metabolites with some drugs (e.g., acetaminophen). Drugs that induce enzyme production also may increase the rate of metabolism for endogenous steroidal hormones (e.g., cortisol, estrogens, testosterone, vitamin D). However, enzyme induction does not occur for 1 to 3 weeks after an inducing agent is started, because new enzyme proteins must be synthesized.
Metabolism also can be decreased or delayed in a process called enzyme inhibition, which most often occurs with concurrent administration of two or more drugs that compete for the same metabolizing enzymes. In this case, smaller doses of the slowly metabolized drug may be needed to avoid adverse effects and toxicity from drug accumulation. Enzyme inhibition occurs within hours or days of starting an inhibiting agent. Cimetidine, a gastric acid suppressor, inhibits several CYP enzymes (e.g., 1A, 2C, 2D, 3A) and can greatly decrease drug metabolism. The rate of drug metabolism also is reduced in infants (their hepatic enzyme system is immature), in people with impaired blood flow to the liver or severe hepatic or cardiovascular disease, and in people who are malnourished or on low-protein diets.
When drugs are given orally, they are absorbed from the GI tract and carried to the liver through the portal circulation. Some drugs are extensively metabolized in the liver, with only part of a drug dose reaching the systemic circulation for distribution to sites of action. This is called the first-pass effect or presystemic metabolism.
Excretion
Excretion refers to elimination of a drug from the body. Effective excretion requires adequate functioning of the circulatory system and of the organs of excretion (kidneys, bowel, lungs, and skin). Most drugs are excreted by the kidneys and eliminated (unchanged or as metabolites) in the urine. Some drugs or metabolites are excreted in bile and then eliminated in feces; others are excreted in bile, reabsorbed from the small intestine, returned to the liver (called enterohepatic recirculation), metabolized, and eventually excreted in urine. Some oral drugs are not absorbed and are excreted in the feces. The lungs mainly remove volatile substances, such as anesthetic gases. The skin has minimal excretory function. Factors impairing excretion, especially severe renal disease, lead to accumulation of numerous drugs and may cause severe adverse effects if dosage is not reduced.
NCLEX Success
1. A nurse practitioner (NP) has just changed a patient’s medication from an oral form to a patch formulation to avoid the first-pass effect. The NP has explained it to the patient, but the patient still has questions and asks the nurse to explain again what is meant by the first-pass effect. The NP would be most correct in explaining that this has to do with how
A. drugs initially bind to plasma proteins
B. initial renal function is involved in drug excretion
C. the way drugs first reach their target cells
D. initial metabolism of an oral drug occurs before it reaches the systemic circulation
2. A nurse is reading a research report about use of a medication that describes the pharmacokinetics of a particular medication that a patient is taking. Pharmacokinetics involves
A. drug effects on human cells
B. drug binding with receptors
C. drug absorption, distribution, metabolism, and elimination
D. drug stimulation of normal cell functions
3. A nurse is caring for a man who has worsening liver disease. In monitoring his medication, it is important to know that a patient with liver disease may have impaired drug
A. absorption
B. distribution
C. metabolism
D. excretion
Serum Drug Levels
A serum drug level is a laboratory measurement of the amount of a drug in the blood at a particular time (Fig. 2.5). It reflects dosage, absorption, bioavailability, half-life, and the rates of metabolism and excretion. A minimum effective concentration (MEC) must be present before a drug exerts its pharmacologic action on body cells; this is largely determined by the drug dose and how well it is absorbed into the bloodstream. A toxic concentration is a level at which toxicity occurs; what is toxic for some patients is not toxic for others (see subsequent discussion). Toxic concentrations may stem from a single large dose, repeated small doses, or slow metabolism that allows the drug to accumulate in the body. Between these low and high concentrations is the therapeutic range, which is the goal of drug therapy—that is, enough drug to be beneficial but not enough to be toxic.
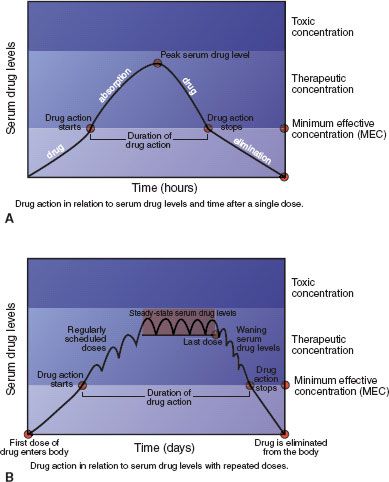
Figure 2.5 Serum drug levels with single (A) and multiple (B) oral drug doses. Drug action starts when enough drug is absorbed to reach the minimum effective concentration (MEC), continues as long as the serum level is above the MEC, wanes as drug molecules are metabolized and excreted (if no more doses are given), and stops when the serum level drops below the MEC. The goal of drug therapy is to maintain serum drug levels in the therapeutic range.
For most drugs, serum levels indicate the onset, peak, and duration of drug action. When a single dose of a drug is given, onset of action occurs when the drug level reaches the MEC. The drug level continues to climb as more of the drug is absorbed, until it reaches its highest concentration and peak drug action occurs. Then, drug levels decline as the drug is eliminated (i.e., metabolized and excreted) from the body. Although there may still be numerous drug molecules in the body, drug action stops when drug levels fall below the MEC. The duration of action is the time during which serum drug levels are at or above the MEC. When multiple doses of a drug are given (e.g., for chronic conditions), the goal is usually to give sufficient doses often enough to maintain serum drug levels in the therapeutic range and avoid the toxic range.
In clinical practice, measuring serum drug levels is useful in several circumstances:
• When drugs with a narrow margin of safety are given, because their therapeutic doses are close to their toxic doses (e.g., digoxin, aminoglycoside antibiotics, lithium)
• To document the serum drug levels associated with particular drug dosages, therapeutic effects, or possible adverse effects
• To monitor unexpected responses to a drug dose such as decreased therapeutic effects or increased adverse effects
• When a drug overdose is suspected



