Chapter 41 Assessment of the Hematologic System
Safe and Effective Care Environment
Health Promotion and Maintenance
2. Assess the patient’s endurance in performing ADLs.
3. Perform a clinical health history and risk assessment for hematologic function.
4. Teach patients and family members about what to expect during procedures to assess hematologic function.
5. Explain the relationship between hematologic problems and the need for oxygen.
6. Describe the hematologic changes associated with aging.
7. Describe the role of platelets in hemostasis.
8. Interpret blood cell counts and clotting tests to assess hematologic status.
9. Explain the effects of anticoagulants, fibrinolytics, and inhibitors of platelet activity on hematologic function.
10. Prioritize nursing care for the patient after bone marrow aspiration.

http://evolve.elsevier.com/Iggy/
Answer Key to NCLEX Examination Challenges and Decision-Making Challenges
Review Questions for the NCLEX® Examination
The blood, blood cells, lymph, and organs involved with blood formation or blood storage together compose the hematologic system. This system is important for oxygenation and tissue perfusion because the blood itself is one of the oxygen delivery systems (Fig. 41-1). All systems depend on the blood for oxygen perfusion. Thus any problem of the hematologic system affects total body health and well-being. This chapter, together with Chapter 19, reviews the normal physiology of the hematologic system and assessment of hematologic status.
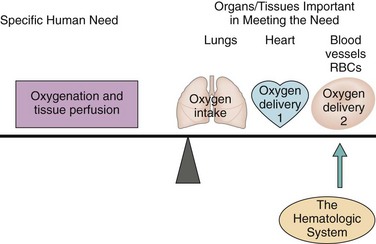
FIG. 41-1 Role of the hematologic system in oxygenation and tissue perfusion. RBCs, Red blood cells.
Anatomy and Physiology Review
Bone Marrow
Bone marrow is the tissue responsible for blood formation. It produces red blood cells (RBCs, erythrocytes), white blood cells (WBCs, leukocytes), and platelets. Bone marrow also is involved in the immune responses (see Chapter 19).
Each day the bone marrow normally releases about 2.5 billion RBCs, 2.5 billion platelets, and 1 billion WBCs per kilogram of body weight. In adults, cell-producing marrow is present only in flat bones (sternum, skull, pelvic and shoulder girdles) and the ends of long bones. With aging, fatty tissue slowly replaces active bone marrow and only a small portion of the remaining marrow continues to produce blood in older adults (Touhy & Jett, 2010).
The bone marrow first produces blood stem cells, which are immature, unspecialized (undifferentiated) cells. These are capable of becoming any one of several types of blood cells: RBCs, WBCs, or platelets, depending on the body’s needs (Fig. 41-2) (McCance et al., 2010).
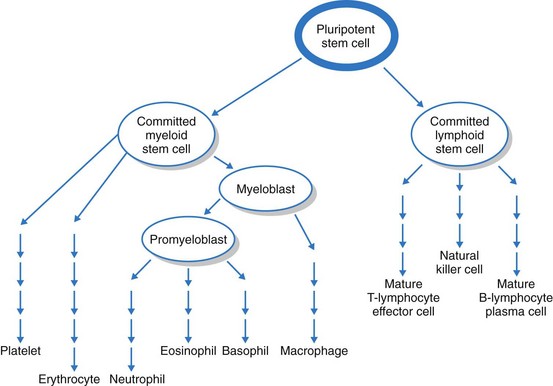
The next stage in blood cell production is the committed stem cell (also called the precursor cell). A committed stem cell enters one growth pathway and can at that point specialize (differentiate) into only one cell type. Committed stem cells actively divide but require the presence of a specific growth factor for specialization. For example, erythropoietin is a growth factor made in the kidneys that is specific for the RBC. Other growth factors influence WBC and platelet growth (see Chapters 19, 24, and 42 for discussion of growth factors and cytokines).
Blood Components
Albumin maintains the osmotic pressure of the blood, preventing the plasma from leaking into the tissues (see Chapter 13). Globulins have many functions, such as transporting other substances and, as antibodies, protecting the body against infection. Fibrinogen is an inactive protein that is activated to form fibrin. Fibrin molecules assemble together to form structures important in the blood clotting process.
As shown in Figs. 41-2 and 41-3, RBCs start out as stem cells, enter the myeloid pathway, and progress in stages to mature RBCs (erythrocytes). Healthy, mature RBCs have a life span of about 120 days after being released into the blood. As RBCs age, their membranes become more fragile. These old cells are trapped and destroyed in the tissues, spleen, and liver. Some parts of destroyed RBCs (e.g., iron, hemoglobin) are recycled and used to make new RBCs.
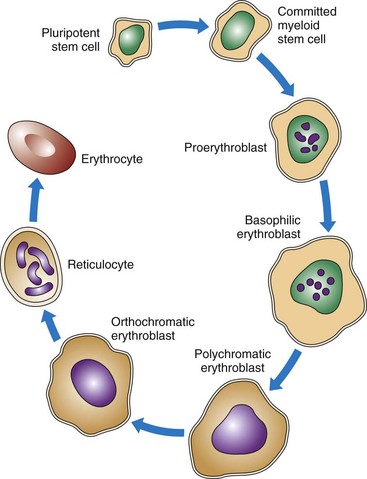
The most important feature of hemoglobin is its ability to combine loosely with oxygen. With only a small drop in tissue oxygen levels, an increase in the transfer of oxygen from hemoglobin to tissues occurs, known as oxygen dissociation. See Chapter 29 for a complete discussion of the physiological concept of oxygen dissociation.
White blood cells (WBCs, leukocytes) perform actions important for protection through inflammation and immunity (Table 41-1). The many types of WBCs all have specialized functions. Many WBCs are formed in the bone marrow and are part of the hematologic system. WBC function is presented in Chapter 19.
TABLE 41-1 FUNCTIONS OF SPECIFIC LEUKOCYTES
| LEUKOCYTE | FUNCTION |
|---|---|
| Inflammation | |
| Neutrophil | Nonspecific ingestion and phagocytosis of microorganisms and foreign protein |
| Macrophage | Nonspecific recognition of foreign proteins and microorganisms; ingestion and phagocytosis |
| Monocyte | Destruction of bacteria and cellular debris; matures into macrophage |
| Eosinophil | Weak phagocytic action; releases vasoactive amines during allergic reactions |
| Basophil | Releases histamine and heparin in areas of tissue damage |
| Antibody-Mediated Immunity | |
| B-lymphocyte | Becomes sensitized to foreign cells and proteins |
| Plasma cell | Secretes immunoglobulins in response to the presence of a specific antigen |
| Memory cell | Remains sensitized to a specific antigen and can secrete increased amounts of immunoglobulins specific to the antigen |
| Cell-Mediated Immunity | |
| T-lymphocyte helper/inducer T-cell | Enhances immune activity through the secretion of various factors, cytokines, and lymphokines |
| Cytotoxic-cytolytic T-cell | Selectively attacks and destroys non-self cells, including virally infected cells, grafts, and transplanted organs |
| Natural killer cell | Nonselectively attacks non-self cells, especially body cells that have undergone mutation and become malignant; also attacks grafts and transplanted organs |
Hemostasis/Blood Clotting
Intrinsic factors are conditions directly in the blood itself that first activate platelets and then trigger the blood clotting cascade (Fig. 41-4). These conditions include circulating debris and prolonged venous stasis. Continuing the cascade to the point of blood clotting requires having sufficient amounts of all the different clotting factors and cofactors (Table 41-2).
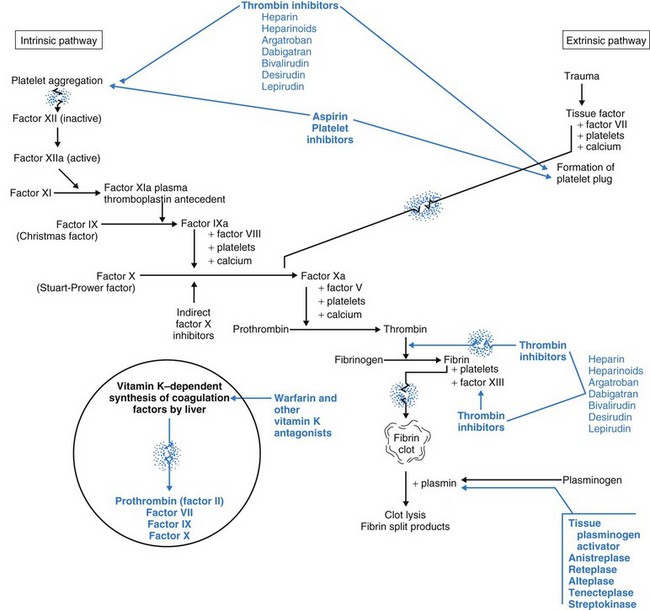
TABLE 41-2 THE CLOTTING FACTORS
| FACTOR | ACTION |
|---|---|
| I: Fibrinogen | Factor I is converted to fibrin by the enzyme thrombin. Individual fibrin molecules form fibrin threads, which are the scaffold for clot formation and wound healing. |
| II: Prothrombin | Factor II is the inactive precursor of thrombin. Prothrombin is activated to thrombin by coagulation factor X (Stuart-Prower factor). After it is activated, thrombin converts fibrinogen (coagulation factor I) into fibrin and activates factors V and VIII. Synthesis is vitamin K–dependent. |
| III: Tissue thromboplastin | Factor III interacts with factor VII to initiate the extrinsic clotting cascade. |
| IV: Calcium | Calcium (Ca2+), a divalent cation, is a cofactor for most of the enzyme-activated processes required in blood coagulation. Calcium also enhances platelet aggregation and makes red blood cells clump together. |
| V: Proaccelerin | Factor V is a cofactor for activated factor X, which is essential for converting prothrombin to thrombin. |
| VI: Discovered to be an artifact | No factor VI is involved in blood coagulation. |
| VII: Proconvertin | Factor VII activates factors IX and X, which are essential in converting prothrombin to thrombin. Synthesis is vitamin K–dependent. |
| VIII: Antihemophilic factor | Factor VIII together with activated factor IX enzymatically activates factor X. In addition, factor VIII combines with another protein (von Willebrand’s factor) to help platelets adhere to capillary walls in areas of tissue injury. A lack of factor VIII is the basis for classic hemophilia (hemophilia A). |
| IX: Plasma thromboplastin component (Christmas factor) | Factor IX, when activated, activates factor X to convert prothrombin to thrombin. This factor is essential in the common pathway between the intrinsic and extrinsic clotting cascades. A lack of factor IX causes hemophilia B. Synthesis is vitamin K–dependent. |
| X: Stuart-Prower factor | Factor X, when activated, converts prothrombin into thrombin. Synthesis is vitamin K–dependent. |
| XI: Plasma thromboplastin antecedent | Factor XI, when activated, assists in the activation of factor IX. However, a similar factor must exist in tissues. People who are deficient in factor XI have mild bleeding problems after surgery but do not bleed excessively as a result of trauma. |
| XII: Hageman factor | Factor XII is critically important in the intrinsic pathway for the activation of factor XI. |
| XIII: Fibrin-stabilizing factor | Factor XIII assists in forming cross-links among the fibrin threads to form a strong fibrin clot. |
Clotting factors (see Table 41-2) are inactive enzymes that become activated in a sequence. The last part of the sequence is the activation of fibrinogen into fibrin. At each step, the activated enzyme from the previous step activates the next enzyme. The last two steps in the cascade are the activation of thrombin from prothrombin and the conversion (by thrombin) of fibrinogen into fibrin. Only fibrin molecules can begin the formation of a true clot.
 NCLEX Examination Challenge
NCLEX Examination ChallengeAnti-Clotting Forces
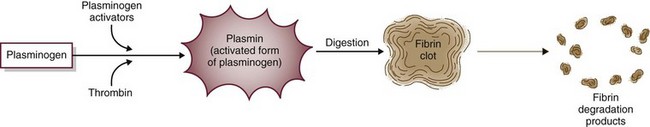
Fibrinolysis limits the size of blood clots by dissolving fibrin clot edges with special enzymes (Fig. 41-5). The process starts by activating plasminogen to plasmin. Plasmin, an enzyme, then digests fibrin, fibrinogen, and prothrombin, controlling the size of the fibrin clot.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


