CHAPTER 35 1. Describe assessment strategies for diagnosis of infants experiencing a congenital defect. 2. Identify methods of initial management and care for individual congenital anomalies. 3. List possible causative factors that result in common congenital abnormalities. 4. Verbalize the importance of parental involvement in the development of infants affected with congenital anomalies. In North America, congenital malformations account for more than 20% of infant deaths (Moore and Persaud, 2013). In the United States, nearly 1 in 120 live-born infants will experience a chromosomal abnormality (Moore and Persaud, 2013). These statistics represent a significant challenge to health care providers and pose life-changing challenges for the families of these infants. A congenital anomaly is defined as a physical, metabolic, anatomic, or behavioral deviation from the normal pattern of development (Moore and Persaud, 2013) (Box 35-1). This chapter presents information concerning the incidence, etiology, clinical presentation, and treatment modalities for some common abnormalities. The reader is directed to Chapter 20 for a foundation on genetics. 1. The frequency of medically significant malformations diagnosed in the newborn period is documented as 2% of all live births (Parikh and Wiesner, 2011). These defects present as single or multiple defects, all with varying clinical significance. a. By 1 year of age, the incidence of congenital anomalies requiring surgery or interfering with normal functioning ranges from 4% to 7% (Lashley, 2005). b. Minor single anomaly rates are estimated to be as high as 14% (Moore and Persaud, 2013). Infants experiencing three or more minor abnormalities commonly have one or more major defects. c. The identification of neonates with a major malformation is vital as these infants have a five-fold increase in morbidity (Parikh and Wiesner, 2011). 2. Birth defects are a significant cause of miscarriage and fetal death. b. Fifty percent to 60% of all infants spontaneously aborted will have a detectable chromosomal abnormality (Lashley, 2005). Of those infants, trisomic abnormalities account for 50% of pregnancy losses (Parikh and Weisner, 2011). B. Etiology. The etiology of 50% to 60% of congenital anomalies is unknown (Moore and Persaud, 2013). Congenital malformations may have more than one cause, are often associated with multiple anomalies of major or minor significance, and have variable recurrence risks. Congenital anomalies may result from chromosomal disorders, single-gene defects, infective agents, teratogens, combinations of genetic and environmental factors, maternal metabolic factors, and mechanical constraints on the uterus (Lashley, 2005). a. Genetic factors are numerically the most frequent cause of congenital anomalies and are responsible for approximately 0.5% to 0.7% of all anomalies (Matthews and Robin, 2011). Chromosomal disorders account for 10% of all of the major malformations (Parikh and Wiesner, 2011). Of anomalies with known causes, 85% are due to genetic factors (Moore and Persaud, 2013). b. Chromosomal errors are produced in the germline of either parent, secondary to errors in fertilization, meiosis, or mitosis (commonly nondisjunction and anaphase lag), and chromosome breakage and reunion (Lashley, 2005). c. Embryos resulting from these unions display missing or extra chromosomes (numeric abnormality) or rearranged segments (structural abnormalities). Chromosomal aberrations result in defective zygotes, blastocysts, and early embryos. d. Both sex chromosomes and/or autosomes may be affected. e. Categories of genetic factors: (1) Numeric abnormalities commonly result from nondisjunction of genetic material. This error in cell division occurs during mitosis or meiosis when a chromosomal pair or two chromatids of a chromosome fail to disjoin (Moore and Persaud, 2013). The end result is the loss or gain of one or more chromosomes (Matthews and Robin, 2011). (b) Monosomy: zygote develops missing a specific chromosome. (c) Mosaicism: Results secondary to nondisjunction in early cleavage division of a single zygote. The result is an embryo having two or more cell lines with different cell numbers (Moore and Persaud, 2013). Symptoms are generally less severe than if all cells are affected. f. Numeric chromosomal abnormalities are frequently associated with intrauterine growth retardation, dysmorphic features, mental retardation, and physical malformations (Matthews and Robin, 2011). g. Structural abnormalities frequently result from chromosome breakage that results in a loss or rearrangement of the broken segment to a different location on the chromosome or to a different chromosome. The resulting change in structure is dependent on the final disposition of the broken pieces. Chromosome breakage may be caused by radiation, drugs, chemicals, and viruses (Moore and Persaud, 2013). (2) Duplication anomalies result as a duplication of genetic material within, attached to, or as a separate fragment of the chromosome (Moore and Persaud, 2013). Though there is no loss of genetic material, individuals commonly experience mental retardation or other birth defects. (3) Inversion defects result when a chromosome segment is reversed. Risk for abnormality is dependent on the portion of the chromosome involved. h. Single-gene or Mendelian disorders are responsible for 4% of major malformations (Parikh and Wiesner, 2011). The mode of Mendelian inheritance for many major malformations is autosomal dominant (Parikh and Wiesner, 2011). Autosomal recessive, or less frequently X-linked, inheritance accounts for a minority of major malformations. The mechanism for development of abnormality is related to dysfunction of the gene or disturbance of the developmental pathway. i. Multifactorial disorders result secondary to interaction of genes and additive environmental influences. In order for an abnormality to be expressed, a threshold of traits must be exceeded. j. Disorders are familial, but lack the inheritance traits of single-gene defects. k. Most isolated single malformations, such as congenital heart defects, neural tube defects, and cleft lip and palate, develop secondary to multifactorial inheritance patterns (Matthews and Robin, 2011). l. Recurrence rates are higher for first-degree relatives, are greater if a larger number of family members are affected, and are increased if the malformation is severe (Matthews and Robin, 2011). 2. Disorders resulting from exposure to teratogens that affect the developing fetus. a. A teratogen is defined as any organism, substance, deficiency state, or physical agent capable of inducing abnormal structure or function (Blackburn, 2013). b. Environmental factors are causative for 7% to 10% of congenital anomalies (Moore and Persaud, 2013). The exact mechanism resulting in a disruption of embryonic development and induction of anomalies remains unclear, but environmental factors do not appear to exert effect until cellular differentiation begins. c. The majority of teratogenic agents exert their effect by interfering with cellular metabolic activity. The end result is failure of cellular replication, cell migration, cellular fusion, and death. Moore and Persaud (2013) define the most critical periods of development as the time when cell division, cell differentiation, and morphogenesis are at their peak. d. The ability of a particular agent to exert a teratogenic effect is determined by the timing of the exposure (critical time once cellular differentiation has begun), dosage of the teratogen (higher the dose, greater the effect), the individual properties of the teratogen, and the genetic susceptibility of the mother and fetus (Matthews and Robin, 2011). e. The range of teratogenic agents is large and continues to increase. McLean (2005) describes classifications of teratogens as infectious agents, drugs and chemical agents, radiation, and maternal factors, as well as mechanical forces upon the fetus. f. Exposure of the fetus to alcohol carries significant risk throughout pregnancy and is considered to be the most common teratogen (Parikh and Wiesner, 2011). Fetal alcohol spectrum disorder is estimated to affect as much as 1% of the general population (Moore and Persaud, 2013). C. Evaluation. When a newborn is identified as having one or more malformations, a detailed history and physical examination is needed to aid in accurate diagnosis. 2. A pedigree analysis should be developed. a. Reproductive losses and infertility. b. Relatives with mental retardation or known malformations. c. Infants within the family with malformations or birth defects. d. Neonatal deaths, childhood deaths, stillbirths. e. Familial disorders or physical features common to the family. f. Consanguinity in parents. g. Ethnic background. 3. Prenatal and perinatal history should detail information about maternal and fetal well-being. a. Maternal age, parity, and health—including maternal illness and medications. b. Pregnancy mode (natural or assisted), complications. c. Teratogenic exposures, including alcohol, drugs, herbal preparations, medications, bacterial infections, viral infections, and parasitic infections. d. Duration of pregnancy, intrapartum course, prenatal testing, and duration. e. Fetal growth and behavior in utero—patterns of movement throughout pregnancy. f. Delivery mode, complications, condition of infant at delivery. g. Birth weight, length, and head circumference and whether measurements are appropriate for gestation. 4. Detailed physical examination is undertaken with attention to physical variations and malformations. It is essential to determine if an anomaly is isolated or part of a pattern of malformation. Clinical photographs should be obtained. Photographs of unusual features provide a permanent record that allows initial diagnosis and later consultation (Lashley, 2005). b. Estimation of gestational age. c. General appearance: posture, tone, and position. d. Systematic examination of all body surfaces. (1) Head: size measured as occipital–frontal circumference, shape (assess for dolichocephaly, premature fusion of cranial sutures, frontal bossing), size of anterior fontanelles (Haldeman-Englert et al., 2012a). (2) Scalp: hair patterns, including placement of whorls, hairline, texture, pigmented areas, eyebrow length and pattern, eyelash length, widow’s peaks, or alterations in pigmentation. (3) Face: configuration, elfin, coarse, flat, triangular, round, birdlike, expressionless. (4) Eyes: spacing of eyes, interpupillary distances, epicanthal folds, palpebral fissures (length and degree of slanting), iris color, colobomas, ptosis. (5) Nose: appearance—beaked, pinched, upturned, flattened bridge, position on face, number and patency of nares, length of the columella. (6) Mouth: shape of palate and uvula, presence of cleft lip or palate, size of tongue or deformities, natal teeth, deformities of frenulum, shape of mouth, philtrum, and vermilion border. (7) Ears: location, position, rotation, unilateral/bilateral defect, protruding/prominent shape, patency of auditory meatus, preauricular and postauricular pits and tags. (8) Neck: length, webbed/redundant skinfolds, posterior hairline, torticollis, small or receding chin, bony abnormalities of the neck. (9) Chest: shape, size, symmetry; location, spacing, and number of nipples, internipple distance. (10) Cardiovascular: murmurs, pulses, blood pressure. (11) Lungs: equality, character of breath sounds. (12) Abdomen: integrity of abdominal wall, location and appearance of umbilicus, presence of masses, organomegaly, hypoplasia of musculature, omphalocele, gastroschisis. Umbilicus should be examined for number of vessels, hernias (Haldeman-Englert et al., 2012a). (13) Genitalia: presence of ambiguity, size, appearance. (14) Anus: location and patency. (15) Spine: neural tube defects, unusual pigmentary lesions, hair tufts, dimples, sinuses. (16) Extremities: proportions, appearance, range of motion, number and placement of hands, feet, digits. Epidural ridges, creases, absence deformities, polydactyly, syndactyly, contracture deformities, clinodactyly, camptodactyly (Parikh and Wiesner, 2011). (17) Skin: pigmentation, lesions, texture; hair distribution, patterns—whorls, widow’s peaks. 5. Diagnostic evaluation of a congenital abnormality. b. Cytogenic, molecular, and biochemical examination are useful in determining etiology of congenital anomalies. c. Chromosome analysis should be obtained for all infants having two or more major malformations, infants with growth restriction in association with anomalies, infants having multiple minor abnormalities, and infants with ambiguous genitalia. (1) Karyotype analysis is performed on cells undergoing mitosis (Haldeman-Englert et al., 2012a). As chromosomes condense, they are stained and a representation of chromosome number results. Normal karyotype consists of 46 chromosomes—22 pairs of autosomes and one set of sex chromosomes. d. Microarray technology allows for detection and analysis of thousands of genes at the same time. Assay provides information about patterns of gene expression and interaction (Haldeman-Englert et al., 2012a). The focus of microarray-based techniques is centered on examination of chromosome number changes. Diagnostic confirmation of microdeletion error is possible. Comparative genomic hybridization, single-nucleotide polymorphism or oligonucleotide arrays, and molecular analysis are being utilized in greater frequency to determine etiology of abnormalities (Haldeman-Englert et al., 2012a). (1) Fluorescent in-situ hybridization (FISH) utilizes segments of fluorescently labeled DNA probes that attach to a specific segment of the chromosome and appear fluorescent under microscopic evaluation. Where material is missing from the segment, the probe is unable to attach to the chromosome, resulting in identification of microdeletion (Murray, 2012). e. Biochemical studies are performed on ill neonates who have a condition that may be secondary to an inborn error of metabolism, or for whom a specific diagnosis cannot be made. Laboratory studies should be obtained before treatment is begun. Presenting symptoms guide the specific tests to be ordered. Newborn screening using tandem mass spectrometry allows for a single drop of blood to effectively screen for 40 inborn errors of metabolism. f. Ophthalmologic examination may be useful as abnormalities are often associated with neurologic or brain malformations. D. Genetic counseling. a. Genetic counseling is offered to prospective parents to help them evaluate risks for hereditary or genetic conditions based on the individual family pedigree (Parikh and Wiesner, 2011). b. Goals of counseling are centered around provision of information concerning diagnosis, needed care, impact of the illness, prediction for recurrence, and future support. Counseling is aimed toward assisting parents to make informed decisions. c. Counseling can be offered by family physicians, neonatologists, or genetic specialists. Counseling is provided in a quiet location with all family members desired by the family present. d. Principles of genetic counseling. (1) Supportive, nonjudgmental attitude. (2) Respectful of family privacy, confidentiality. (3) Sensitive to ethnic, cultural, and language differences. (4) Supportive of family’s movement through the grieving process. (5) Content of counseling should take into account medical issues, mechanisms causing the abnormality, a realistic prognosis, and a plan for treatment and support resources. (6) Risk for recurrence and reproductive options. (7) Assistance with family adjustment and social services as needed. See Figure 35-1 for the division of congenital anomalies. 1. Most common autosomal chromosomal abnormality in live-born infants; occurring in 1 in 800 infants (Haldeman-Englert et al., 2012b). 2. Full trisomy 21 occurs in 94% of cases (all or large part of the chromosome). Affected individuals have 47 chromosomes (three of chromosome 21) (Fig. 35-2). 3. Greater than 90% of trisomy 21 cases result secondary to meiotic disjunction. Maternal age exerts a significant effect. Three percent to 5% of cases are de novo or caused by a balanced translocation that becomes unbalanced (Haldeman-Englert et al., 2012b). 4. Mosaic or mitotic nondisjunction Down syndrome, presenting with variable phenotypic expression, is demonstrated in 3% of affected infants (Haldeman-Englert et al., 2012b). 5. Greater than 50% of trisomy 21 fetuses abort early in pregnancy. 6. Recurrence risk approximately 1% overall. For those parents who are known translocation carriers, the risk is substantially higher. Risk for carrier parent is dependent on the type of translocation and the sex of the parent (Jones et al., 2013). 7. Incidence of Down syndrome related to maternal age is as follows (Moore and Persaud, 2013): b. 25 to 29 years—1 in 1100. c. 30 to 34 years—1 in 700. d. 35 years—1 in 350. e. 37 years—1 in 225. f. 41 years—1 in 85. g. 43 years—1 in 50. h. Over 45 years—1 in 25. B. Clinical presentation. a. Brachycephaly with flattened occiput, presence of third fontanelle. b. Upslanting palpebral fissures, iris speckled with Brushfield’s spots, colobomatous cataracts, and glaucoma. c. Prominent epicanthal folds. d. Flattened facial profile, micrognathia, flattened nasal bridge. e. Small, posteriorly rotated, rounded ears; low-set and malformed ears (overfolding of superior helices), hearing loss. f. Prominent, protruding tongue; high-arched palate with tendency to keep mouth open (Jones et al., 2013). 2. Musculoskeletal. a. Hypotonia: generalized—poor Moro reflex present in 80% of infants. Suck may be weak. b. Hyperflexibility of joints, dysplasia of pelvis: narrow acetabular angle. c. Clinodactyly of fifth digits, single or bilateral transverse simian crease—present in 50% of cases. Brachydactyly. d. Prenatal growth restriction, short stature, often 10% to 25% of affected children. e. Wide spacing between first and second toes (Matthews and Robin, 2011). 3. Skin. a. Short neck with excess nuchal skin. C. Associated findings. 2. Mental retardation: present in all cases, variable degree, mild to moderate impairment (Lashley, 2005). 3. Hematologic: bone marrow dyscrasias, including neonatal thrombocytopenia and transient self-resolving myeloproliferative disorders, have been described within the first year of life (Haldeman-Englert et al., 2012b). Thirty percent of infants with Down syndrome and transient myeloproliferative disease develop acute megakaryoblastic leukemia within the first 4 years of life (Luchtman-Jones et al., 2011). 4. Gastrointestinal: duodenal atresia/stenosis—occur in 2% to 5% of cases; esophageal atresia, imperforate anus, fistulas, webs. 5. Cardiac anomalies in approximately 50% of infants: endocardial cushion defect, ventral septal defect, patent ductus arteriosus, atrial septal defect, tetralogy of Fallot (Haldeman-Englert et al., 2012b). 6. Otitis media, hearing impairment. D. Diagnosis. 1. Chorionic villus sampling at 9 to 12 weeks of gestation. 2. Amniocentesis at 16 weeks. 3. Maternal serum screening for low values of α-fetoprotein (AFP). 4. Ultrasound targeting growth, polyhydramnios, heart defects. 5. Standard chromosome banding techniques provide confirmation of trisomy 21 and differentiate those infants having nondisjunctional trisomy from a translocation. In those infants with confirmed translocation, each parent should have karyotype analysis done to rule out carrier status as recurrence risk is significantly increased (Lewanda et al., 2006). 6. FISH: region on chromosome affected is q22.2. E. Treatment. 1. Initial treatment geared to expressed symptoms. 2. Parent education and support are essential for treatment of continuing health issues. 3. Ophthalmologic evaluation should be undertaken as children with Down syndrome have greater incidence of cataracts, strabismus, myopia, and glaucoma (Haldeman-Englert et al., 2012b). 4. Monitor cerebral spine for spinal cord compression. 5. Monitor growth as velocity often decreases—weight and head circumference. 6. Screen for hypothyroidism. 7. Upper respiratory infections and ear infections are common. 8. Cardiac sequelae, including congestive heart failure. 9. Aimed at optimizing potential for intellectual and social growth. 10. Developmental delays—muscle tone increases with age, rate of developmental progress slows with age (Jones et al., 2013). Early developmental enrichment programs seem to be of the most value. 11. Social performance is usually above the expected level. Emotional problems are apparent in many affected individuals. Parents must be provided with resources for emotional and medical support. 12. Following correction of associated congenital malformations, less than 50% of individuals with Down syndrome survive to age 60. Less than 15% survive to age 68 (Haldeman-Englert et al., 2012b). 1. Affected infants have 47 chromosomes (three of chromosome 18) (Fig. 35-3). 2. Occurs in 1 in 600 births; females more often than males (3:1) (Jones et al., 2013). Trisomy 18 is the third most common autosomal disorder. Associated with a high rate of intrauterine demise—5% of affected cases will survive to birth (Haldeman-Englert et al., 2012b). 3. Eighty percent of cases are caused by chromosomal nondisjunction (Lewanda et al., 2006); 95% of infants have three copies of entire chromosome 18, and 5% have either partial trisomy or mosaicism of most of the long arm of 18 (Haldeman-Englert et al., 2012b). 4. Occurs more frequently as maternal age advances: mean maternal age is 32 (Jones et al., 2013). 5. Three patterns of presentation have been described: a. Severe: most body cells display abnormality. Handicaps are severe, with a short life expectancy. b. Mosaic form: some cells have the normal complement of genetic material, the remaining cells having the typical pattern for trisomy 18. Infants are less severely affected and have a longer life expectancy. c. Partial: dependent on the portion of the chromosome affected. Infants with trisomy of the short arm of the chromosome have minimal handicaps and few abnormalities are expressed. Infants with trisomy involving the entire long arm of the chromosome display the full spectrum of the syndrome. Infants with trisomy of the distal one third of the chromosome demonstrate a partial syndrome with less profound mental deficit and longer survival (Jones et al., 2013). 6. Estimate for risk of recurrence for future pregnancies is 1% over the maternal age-specific risk for any viable autosomal trisomy (Haldeman-Englert et al., 2012b). Infants whose defect is secondary to a structural arrangement warrant parental karyotype analysis. B. Clinical presentation. 1. General: growth deficiency, hypoplasia of skeletal muscle, subcutaneous and adipose tissue (Haldeman-Englert et al., 2012b). 2. Craniofacial: small narrow cranium, prominent occiput, low-set malformed auricles, atresia of auditory canals, narrow palpebral fissures, microphthalmia, corneal opacities, colobomas, micrognathia, microstomia and high-arched palate, cleft palate (Lewanda et al., 2006). 3. Musculoskeletal: clenched hand, with index finger overlapping the third finger and the fifth finger overlapping the fourth, abnormal creases, low-arch dermal ridge pattern on six or more fingertips, hypoplasia of nails, short sternum, narrow pelvis with hip dislocation, rocker-bottom appearance to feet, hammer toes, clubfeet, and syndactyly between toes 2 and 3 (Jones et al., 2013). 4. Skin: redundant skin, mild hirsutism of back and forehead. C. Associated findings. 1. Cardiac anomalies: varied. Occur in 95% of cases (Lashley, 2005). Ventricular septal defects and patent ductus arteriosus, atrial septal defects, pulmonic stenosis, coarctation of the aorta are most common (Haldeman-Englert et al., 2012b). 2. Renal anomalies: horseshoe kidneys, ectopic kidneys, double ureters, and cystic kidneys. 3. Genital abnormalities: cryptorchidism in males. Hypoplasia of labia and prominent clitoris in females. 4. Umbilical hernias. 5. Severe psychomotor retardation. D. Diagnosis. 1. Chorionic villus sampling at 9 to 12 weeks of gestation. 2. Amniocentesis at 16 weeks. 3. Maternal serum triple screening revealing low levels of AFP, unconjugated estradiol, and total human chorionic gonadotropin. Fifty percent to 60% of trisomy 18 cases can be detected with second-trimester screening utilizing the triple screen (Blackburn, 2013). 4. Ultrasound targeting growth retardation, oligohydramnios and polyhydramnios, heart defects, myelomeningocele, and limb abnormalities. 5. Following delivery, standard chromosome banding assay or FISH. E. Treatment. 1. Infants with full trisomy 18 are usually fragile, with a history of feeble fetal activity (Jones et al., 2013). Prognosis for infants with this disorder is poor. More than 90% of infants will succumb in the first 6 months of life, 5% will survive to 1 year (Haldeman-Englert et al., 2012b). 2. Children who survive the first year of life deal with feeding issues secondary to hypotonia, growth issues, and significant developmental disability. These children generally do not walk unsupported or develop expressive language, but can be capable of limited verbal communication and some social interaction. 3. Once a diagnosis has been confirmed, decisions about extraordinary medical means for prolongation of life present nearly overwhelming challenges for families. Individual circumstances for each family must be considered (Jones et al., 2013). Most deaths result secondary to congestive heart disease, infection, and central apnea (Haldeman-Englert et al., 2012b). 4. For those infants surviving the immediate newborn period, care is supportive and individualized to the infant’s needs. 5. Parents should be offered genetic counseling and provided with resources for support. 1. Affected infants are born with 47 chromosomes instead of the usual 46, with the extra genetic material located on chromosome 13. This abnormality is the rarest of the trisomies, severe external malformations are common, and only 2% to 3% survive to term (Lashley, 2005). 2. Defect occurs in 1 in 12,500 to 1 in 21,000 live births (Haldeman-Englert et al., 2012b), making it the fourth most common autosomal dominant disorder. a. Older mothers are more likely to have an infant with this aneuploidy defect. b. Slightly more boys than girls are affected (Jones et al., 2013). 3. Seventy-five percent of cases result from chromosomal nondisjunction (Lewanda et al., 2006). 4. The etiology for this disorder is trisomy of all or part of chromosome 13. a. Eighty percent of affected infants have three complete copies of chromosome 13. b. Mosaicism accounts for 5% of cases, with these infants demonstrating a wide range of expression from near normal to severe full pattern malformation. (1) Survival for infants with mosaicism is usually longer. (2) Degree of mental deficit is variable (Jones et al., 2013). c. Partial trisomy of the proximal segment is characterized by infants with severe mental deficiency and an overall pattern of full trisomy 13. d. Partial trisomy of the distal segment results in severe mental deficiency and characteristic phenotype (Jones et al., 2013). B. Clinical presentation. 1. General: growth deficiency. 2. Craniofacial: cebocephaly, microcephaly, microphthalmia, colobomata, glaucoma, midline scalp defects, retinal dysplasia, cleft lip and/or palate (60% to 80%) (Jones et al., 2013), malformed ears, atresia of external auditory canals, deafness, prominent nasal bridge, short neck with excessive skin. a. Aplasia cutis congenita is a congenital localized absence of skin often involving the scalp along the midline. Lesions have sharp margins and may present as ulcers, bullae, or scars (Tran and Cohen, 2012). Lesions are frequently noted on the scalp near the parietal hair whorl (Graham, 2007). Most defects are small and superficial; 20% involve absence of the skull, exposing the infant to hemorrhage and infection. 3. Musculoskeletal: polydactyly, single palmar crease, overlapping of fingers, flexion deformities of hands and wrists, prominent heel resulting in rocker-bottom feet, thin posterior ribs with or without missing rib, and hypoplasia of pelvis with shallow acetabular angle. 4. Genital abnormalities: males—cryptorchidism, abnormal scrotum; females—bicornuate uterus. 5. Central nervous system (CNS): holoprosencephaly found in 50% of patients with incomplete development of the forebrain and olfactory and auditory nerves, seizure activity, apnea in early infancy, and severe mental deficiency (Haldeman-Englert et al., 2012b). 6. Capillary hemangiomas, localized scalp defects—cutis aplasia. C. Associated findings. 2. Renal abnormalities: polycystic kidneys. 3. Inguinal or umbilical hernias, single umbilical artery. D. Diagnosis. 1. Chorionic villus sampling between 9 and 12 weeks of pregnancy. 2. Amniocentesis at 16 weeks. 3. Ultrasound targeting growth parameters, abnormalities of heart, kidneys, and brain. 4. Following delivery, standard chromosome banding assay, FISH. E. Treatment. 1. The presence of holoprosencephaly is the single most important finding predicting survival. 2. Prognosis is extremely poor—80% mortality in the newborn period has been documented (Haldeman-Englert et al., 2012b). Survivors frequently have seizures, severe mental retardation, and failure to thrive. 3. Feeding issues are related to the smallness of the lower jaw and poor muscle tone. 4. For those infants with cleft lip and/or palate, feeding issues present additional challenges. 5. The high rate of early mortality for this syndrome requires the parents to prepare for the infant’s death as well as plan for supportive care. The desires of the parents must be taken into account as care is designed for each patient. This syndrome has been referred to as DiGeorge syndrome, Sprintzen syndrome, or velocardiofacial syndrome. It is the most commonly occurring microdeletion syndrome in humans (Haldeman-Englert et al., 2012b). 1. Occurs in 1 in 3000 infants (Haldeman-Englert et al., 2012b). This syndrome is extremely variable in expression and varies from patient to patient. 2. Inherited as an autosomal dominant pattern. 3. Individuals have an interstitial deletion of 22q11.2; 80% to 90% of patients have the same large deletion (Haldeman-Englert et al., 2012b). 4. Most 22q11 deletions occur as de novo lesions—less than 10% are inherited from an affected parent. Both parents must be tested to determine carrier status (Jones et al., 2013). B. Clinical presentation. 1. Growth: postnatal onset of short stature (Jones et al., 2013). 2. Craniofacial: microcephaly, velopharyngeal insufficiency, palatal anomalies, small or absent adenoids, prominent nose with bulbous tip, narrow palpebral fissures, long face, retruded mandible with chin deficiency, small mouth—associated with swallowing abnormalities, hooded eyelids, hypertelorism, hearing deficits, and minor auricular abnormalities. 3. Musculoskeletal: limbs are slender, hypotonic; hands and fingers are hyperextensible (Jones et al., 2013). C. Associated findings. 1. Cardiac defects are present and usually significant in 85% of patients. Most common defects include ventral septal defects, right interrupted aortic arch, tetralogy of Fallot, and truncus arteriosus (Jones et al., 2013). 2. Atresia or hypoplasia of the thymus and parathyroid glands (Haldeman-Englert et al., 2012b). 3. Functional T-cell abnormalities. 4. Hypocalcemia: occurs as a transient finding in neonates (60%). Seizures can occur—usually as a result of hypocalcemia. 5. Learning disabilities: 62% of patients will have normal or mild learning issues, with severe or moderate learning difficulties in 18% (Jones et al., 2013). Developmental delays in language and speech are common (Haldeman-Englert et al., 2012b). 6. IQ range variable. One third of patients function within normal range. 7. Psychiatric disorders—schizophrenia. 8. Speech and language are often delayed. Speech pattern is frequently nasal secondary to poor pharyngeal musculature. D. Diagnosis. 1. Defect is detected by FISH. E. Treatment. 1. Death primarily secondary to cardiac defects—occurs in approximately 8% of infants (Jones et al., 2013). 2. Hypotonia is common, resulting in developmental delays. Early intervention, physical and speech therapy are helpful, especially with feeding issues. 3. Infants may require surgical intervention for palatal abnormalities to improve speech. 4. Infants frequently demonstrate greater socialization skills than intellectual skills. 5. Onset of psychiatric symptoms is usually between 10 and 21 years of age—schizophrenia and depression are the most common conditions. Ongoing assessment and support are essential (Jones et al., 2013). 1. Affected individuals have monosomy—loss of all or part of one copy of the X chromosome in a female. Most 45,X conceptuses die early—0.1% survive to term, the majority are spontaneously aborted (Haldeman-Englert et al., 2012b). a. Loss of the entire chromosome occurs in approximately 50% of cases (Lewanda et al., 2006). 2. Occurs in approximately 1 in 2500 live-born phenotypic females (Haldeman-Englert et al., 2012b). 3. In 70% to 80%, the paternal X chromosome is missing (Lashley, 2005). 4. Mosaicism is associated with complex karyotypes, ring chromosomes, and deletions. 5. Disorder generally occurs as a sporadic event in a family (Jones et al., 2013). B. Clinical presentation. 2. Craniofacial: narrow palate, low-set ears with anomalous auricles, hearing loss, ptosis of eyelids, epicanthal folds, broad nasal bridge, low posterior hairline with appearance of a webbed neck. 3. Musculoskeletal: broad chest with wide-spaced nipples, pectus excavatum, short fourth metacarpal/metatarsal, marked lymphedema of extremities, bone dysplasia, dislocation of the hip, knee abnormalities (Lewanda et al., 2006). 4. Skin: excessively pigmented nevi, loose skin—especially posterior folds of neck. 5. Lymphatic: Impaired lymph drainage leads to distention of lymphatics, and peripheral lymphedema, particularly of dorsa of hands and feet (Kalhan and Devaskar, 2011). C. Associated anomalies. 1. Cardiac defects: bicuspid aortic valve, coarctation of aorta, valvular aortic stenosis, and mitral valve prolapse (Haldeman-Englert et al., 2012b). 2. Renal: horseshoe kidney, unilateral renal agenesis. 3. Gonads: ovarian dysgenesis with hypoplasia owing to lack of germinal elements (Jones et al., 2013). 4. Performance issues: delayed motor skills, poor coordination, clumsiness, problems with nonverbal problem solving, depression and poor self-esteem in young adults (Jones et al., 2013). D. Diagnosis. 1. Ultrasound for short stature, monitoring of growth throughout gestation. 2. Standard chromosome banding assay or FISH. E. Treatment. 1. Growth issues persist throughout childhood and adolescence. Growth is commonly normal within the first 3 years of life, decreasing during school-age years, more dramatically during teenage years when these girls do not get their usual growth spurt at puberty. Growth hormone therapy is generally begun at around 4 to 5 years of age. Significant improvement in final height has been achieved (Haldeman-Englert et al., 2012b). 2. Congenital lymphedema requires symptomatic treatment initially, presenting with edema of hands and feet secondary to faulty lymph drainage. 3. Ovarian function is affected, with primary ovarian failure occurring secondary to gonadal dysplasia. The result can be delay in development of secondary sex characteristics and primary amenorrhea. Treatment options include cyclic estrogen replacement therapy for hypogonadotropic girls beginning at ages 13 to 14 (Haldeman-Englert et al., 2012b). 4. Ongoing evaluation of congenital heart defects is necessary secondary to the increased risk for dissection of the aorta. Coarctation of aorta must be surgically repaired. 5. With increasing age, greater incidences of osteoporosis, autoimmune thyroid disease, and chronic liver disease have been reported (Jones et al., 2013). Close monitoring for these conditions is needed. 6. For those infants for whom their physical appearance would be aided by plastic surgery, concerns for the formation of keloid scars must be considered. 7. School performance may be affected primarily in word comprehension, perceptual thinking, and presentation (Haldeman-Englert et al., 2012b). Intelligence is usually normal. 1. Affects approximately 1 in 1000 males, making it the most common single cause of hypogonadism and infertility in males (Moore and Persaud, 2013). 2. Chromosomal analysis reveals a 47,XXY karyotype. a. Parental nondisjunction errors account for one half of the affected infants; maternal meiosis errors accounting for the majority of remaining cases (Lewanda et al., 2006). b. Advanced maternal age is related to an increased incidence for those whose defect is secondary to maternal factors. c. According to Jones et al. (2013), older fathers are at higher risk for producing a higher number of XY sperm, resulting in a higher risk of having an infant with Klinefelter’s syndrome. 3. Infants with XXY/XY mosaicism may have a greater potential for testicular function. 4. Infants carrying XXYY karyotype are more often developmentally delayed. B. Clinical presentation. 1. General: affected males frequently have long limbs, reduced upper- to lower-segment ratio (Moore and Persaud, 2013). Tall and slim stature. 2. Musculoskeletal: elbow dysplasia, fifth finger clinodactyly. 3. Genital: hypogonadism, cryptorchidism, hypospadias, gynecomastia. C. Associated findings. 1. Cancers: teratoma, leukemia, breast most common. 2. Scoliosis during adolescent years (Jones et al., 2013). 3. School performance and behavioral issues: delayed speech and language development, difficulties with auditory processing and auditory memory. Aggressive behavior is observed (Moore and Persaud, 2013). D. Diagnosis. 1. Prepubertal males have no significant dysmorphism. Condition may not be diagnoses until puberty, with incomplete development of secondary sex characteristics and infertility (Lashley, 2005). Minor abnormalities are helpful in providing clues for diagnosis: penile size, skeletal defects (Lewanda et al., 2006). 2. Chromosomal testing, FISH to determine XXY or XXYY or XXXY status. E. Treatment. 1. As adolescence progresses, screening for scoliosis is needed. 2. Deficient testosterone and elevated gonadotropin levels signal the need for hormone replacement. Boys must be monitored for prospective testosterone replacement as they near ages 10 to 12. As some males have incomplete or inadequate puberty, replacement will assist in development of a more masculine physique, increase pubic and facial hair, and improve bone density and muscle mass (Jones et al., 2013). Delays in treatment can increase risk for osteoporosis. 3. The incidence of breast cancer for affected infants is 20 times higher than the general population, but only 1 in 5000 affected men. Routine mammography is not required (Jones et al., 2013). Incidence of mediastinal teratomas is 34 to 40 times that of nonaffected populations. Ongoing assessment of these individuals is needed. 4. IQ range for affected individuals is variable, from well below to well above average. Assistance in school is commonly required, especially in the areas of reading and spelling. 5. Emotional and behavioral problems persist and may increase throughout childhood. Formation of peer relationships may be difficult secondary to insecurity, shyness, and poor judgment activity. Support with psychological adjustment is needed. Malformation sequences occur with a single, localized, poor formation of tissue that initiates a chain of subsequent defects (Jones et al., 2013). Expressed manifestations range from nearly normal to severe and carry a variable recurrence risk. Clinically and genetically heterogeneous hereditary disease involving connective tissue disorders and skeletal dysplasia. 1. Disorder is caused by mutations of COL1A1 and COL1A2 genes on chromosomes 17q21 and 17q22.1. The result of the mutation is an abnormality of type I collagen (Rimoin and Tiller, 2012). Overall incidence is 3 to 4 per 100,000. 2. Six clinical types are identified: a. Type I: autosomal dominant, resulting from mutations of COL1A1 gene on chromosome 17q21 (Jones et al., 2013). Most common form found in most populations. Characterized by bone fragility with onset of fractures following birth. b. Type II: autosomal dominant, inheritance pattern occurring secondary to a sporadic, dominant mutation in one of the two collagen genes (Jones et al., 2013). (1) Frequency 1 in 20,000 to 60,000 (Rimoin and Tiller, 2012). Lethal in the perinatal period—either as stillborn or secondary to respiratory failure; 80% die within the first month of life. Recurrence risk is described as 6%. c. Type III: primarily an autosomal dominant inheritance pattern, although rare autosomal recessive pattern has been identified (Jones et al., 2013). Progressive and deforming; if not present at birth, fractures and deformations develop in first and second years of life (Rimoin and Tiller, 2012). d. Type IV: autosomal dominant pattern resulting from mutations of the two collagen genes. Significant bone deformities are common. e. Type V: autosomal dominant inheritance pattern that is not associated with collagen type I mutations (Jones et al., 2013). Moderate tendencies to fracture long bones. f. Type VI: pattern of inheritance is unknown. Results from a mineralization defect; fractures occurring later between 4 and 18 months. B. Clinical presentation. a. Growth: near-normal or normal growth pattern. b. Craniofacial: macrocephaly, triangular appearing facies, hearing impairment, altered dentition with hypoplasia of dentin and pulp (Jones et al., 2013). c. Musculoskeletal: postnatal onset of mild limb problems; bowing of femur, tibia, scoliosis, hyperextensibility of joints, and osteopenia. d. Skin: translucent, easy bruising, blue sclerae secondary to partial visualization of the choroid. 2. Type II. a. Growth: short-limbed growth deficiency with prenatal onset, low birth weight. b. Craniofacial: head is soft and boggy, with minimal calvarial bone palpable, large fontanelles, deep blue sclerae, shallow orbits; head assumes a triangular shape (Rimoin and Tiller, 2012). c. Musculoskeletal: shortened and bowed limbs with extra skin, flexed and abducted hips, flattened vertebrae (Jones et al., 2013). 3. Type III. a. Growth: prenatal onset of growth deficiency, extremely short stature. b. Craniofacial: macrocephaly, triangular facies, deep blue sclera, hearing loss, dentinogenesis imperfecta. c. Musculoskeletal: kyphoscoliosis, which may lead to respiratory distress (Jones et al., 2013), multiple fractures, and deformations of limbs at birth. 4. Types IV to VI. C. Associated findings. 1. Conductive hearing loss secondary to deformity of the small bones of the ear. 2. Platelet function is decreased due to defects in adhesion and clot retraction. 3. Corneal clouding and keratoconus, megalocornea (Jones et al., 2013). 4. Cardiac: floppy mitral valve. 5. Scoliosis developing later, progressing during puberty, sometimes resulting in severe deformity in adulthood. Loss of height may occur secondary to progressive spinal osteoporosis in adults. D. Diagnosis. 1. Prenatal diagnosis is possible with ultrasonography and mutation analysis. 2. Radiographs: entire skeleton, including skull, hands, feet, and lateral spine. a. Type I: femurs are short, broad, or crumpled. Fibulas may be thin. b. Type II: femurs are short and deformed, but not crumpled. The long bones appear thin with bowing and deformations. Cranial bones are undermineralized, with wormian bones. 3. Ultrasound of brain, heart, and kidneys. 4. Detailed family history, including measurements of family members. 5. Chromosomal study confirming genetic mutation on 17q21 and 17q22.1. E. Treatment. b. Laxity of ligaments and previous fracture abnormalities complicate the healing process. c. Lightweight splints or braces provide the first-line treatment; depending on previous bone damage, surgical procedures may be necessary to provide strengthening and support. d. Administration of bisphosphonates to increase bone density and decrease fracture has been suggested (Rimoin and Tiller, 2012). 2. Early intervention for hearing loss is essential for development of language and later learning. 3. Dentition may be difficult or delayed; these children are more prone to cavities. Ongoing examination and treatment is vital. 4. Scoliosis may progress quickly for those nearing puberty—bracing is often ineffective. Early surgical fusion may provide the best option for treatment. 5. If a diagnosis has been made prenatally, operative delivery to avoid fractures and intracranial bleeding is recommended. 1. Achondroplasia occurs with a frequency of 1 in 25,000 live births (Rimoin and Tiller, 2012). a. Most common skeletal dysplasia in humans (Lewanda et al., 2006). 2. Defect has an autosomal dominant inheritance pattern. Eighty percent are sporadic occurrences of FGFR3 (fibroblast growth factor receptor 3), representing a new mutation on chromosome 4p16.3 (Rimoin and Tiller, 2012). a. New mutations occur in the father’s sperm and are associated with advanced paternal age (Lewanda et al., 2006). b. Transmitted as a fully penetrant autosomal dominant trait; each person who inherits the mutant gene will show the condition (Rimoin and Tiller, 2012). B. Clinical presentation. 2. Craniofacial: megalocephaly, small foramen magnum, low nasal bridge, flat midface, short flat nose with broad tip, anteverted nares, long philtrum, hypotelorism (Jones et al., 2013). 3. Musculoskeletal: short limbs, lumbar lordosis, mild thoracolumbar kyphosis, short tubular bones, vertebral anomalies, short trident hand, hyperextensible joints, shortened upper limbs, and flexion contractures at elbows. C. Associated findings. 1. Mild hypotonia, especially in the trunk and extremities, with delayed milestones. 2. Bowing of the legs after ambulation has started. 3. Orthodontic problems related to maxillary hypoplasia. 4. Stenosis of the magnum and spine leading to compression of the upper cord and resultant symptoms of apnea, growth delays, hydrocephalus, quadriparesis, and sudden death. 5. Upper airway is often small, leading to obstructive apnea, snoring, and serous otitis media (Rimoin and Tiller, 2012). D. Diagnosis. 1. Chromosomal studies to confirm mutation on 4p16.3 chromosome. 2. Diagnosis is confirmed for achondroplasia through radiographic survey: iliac bones are short and round, lumbar vertebrae have short pedicles and scalloping, long bones have mildly flared metaphyses and are shortened. E. Treatment. 2. Unsupported sitting is associated with development of kyphosis. Infants should not be allowed to be carried in flexed positions before trunk muscle strength is adequate (Rimoin and Tiller, 2012). Most infants lose kyphosis and develop lumbar lordosis when ambulation begins. 3. Hydrocephalus may develop within the first 2 years of life. Radiologic imaging of the skull as a baseline is recommended for monitoring; monthly head circumference should be obtained and plotted to determine abnormal growth. 4. Obstructive apnea may represent a serious threat. Treatment may consist of tonsillectomy or adenoidectomy. Evaluation for foramen magnum stenosis should be done. 5. Treatment for severe bowing of the legs is often deferred until full growth has occurred. Osteotomies provide needed corrections (Jones et al., 2013). 6. Close monitoring of auditory performance is needed as shortened eustachian tubes may lead to middle ear infections and resultant hearing loss. Many infants require placement of tympanic membrane tubes. 7. Dental crowding is frequently encountered, requiring removal of one or more teeth. 8. Obesity develops toward late childhood and should be monitored closely. See Chapter 34. A wide spectrum of abnormalities that affect the femoral head and acetabulum. 1. Defect occurs in 11.5 per 1000 live births, frank dislocations occurring in 1 to 2 per 1000 (White and Goldberg, 2012). Dislocations are described as syndromic or typical. 2. Syndromic dislocations are associated with neuromuscular or dysmorphic conditions. These defects occur in either week 12 or 18 of gestation, and are associated with arthrogryposis and myelodysplasia (White and Goldberg, 2012). 3. Typical dislocations present in healthy infants in prenatal or perinatal period. 4. Risk factors include female gender (19 per 1000), positive family history, and breech presentation. 5. In 60% of infants the left hip is affected, in 20% the right hip is affected, and in 20% both hips (White and Goldberg, 2012). 6. Frank breech positioning with hips flexed and knees bent increases risk of dislocation. 7. Etiology is multifactorial; interaction of genetic and environmental factors is considered to be causative. 8. Ligamentous laxity secondary to circulating maternal hormone (relaxin) exerts an effect, resulting in hip dislocation. Familial ligamentous laxity has also been described. B. Clinical presentation. 1. The defect is considered a deformation anomaly. A full range of severity from a lax dislocatable hip to a nonreducible dislocation (Lashley, 2005). 2. Not all dislocated hips are present at birth. 3. Not all dislocated hips present at birth are present in the newborn period. 4. Despite newborn screening, 1 in 50,000 children will have a dislocated hip detected at 18 months or greater of age (White and Goldberg, 2012). 5. Anatomic changes are minimal in the neonate. As the hip becomes permanently dislocated, pathologic changes of the acetabulum, femoral head, hip capsule joints, and ligaments can occur. End result is degenerative changes in femoral head and acetabulum. 6. Physical examination will demonstrate limitations in adduction, apparent shortening of the affected femur, and extra skinfolds. C. Associated findings. 2. A strong correlation with Larsen’s syndrome (multiple joint dislocations) exists (Jones et al., 2013). 3. Unilateral defects often have secondary problems with limb-length inequality, ipsilateral knee deformity, scoliosis, and disturbances in gait. D. Diagnosis. a. Barlow’s test: attempts to dislocate or subluxate a located, but unstable hip. b. Ortolani test: determines if femoral head is dislocated laterally. c. If the dislocation is a fixed type of lesion, Barlow’s and Ortolani tests are not helpful as the femoral head is locked outside of the acetabulum. Diagnosis is made by palpation of the femoral head posteriorly (Smith, 2012). d. Galeazzi sign: presence of asymmetrical thigh and/or buttock folds. 2. Radiographs are helpful but are difficult to interpret before 4 to 5 months of age secondary to the high percentage of cartilage in newborn infants. a. Ultrasound screening of the cartilaginous femoral head and acetabulum has become common, with sensitivity for detection of over 90% (Lashley, 2005). E. Treatment. 2. Initial goal of treatment is reduction of the displaced femoral head and support of acetabular and femoral head growth. Reduction and stability of the femoral head are needed for normal growth and development of the hip joint. 3. For children 0 to 6 months of age, infants with reducible hip abnormalities are treated with the Pavlik harness (Fig. 35-4). The Pavlik harness is the most commonly used device to prevent adduction while allowing flexion and abduction. Length of treatment is dependent on the age of presentation. The harness encourages deepening and stability of the acetabulum. Progress is judged by serial physical examinations and ultrasonography. Treatment is successful in 90% of patients (White and Goldberg, 2012).
Congenital Anomalies
SPECIFIC DISORDERS
Abnormalities of Chromosomes
Trisomy 21 (Down Syndrome)

Trisomy 18
Trisomy 13
22q11.2 Deletion Syndrome
Sex Chromosome Abnormalities
Turner’s Syndrome
Klinefelter’s Syndrome
NONCHROMOSOMAL ABNORMALITIES
Malformation Disorders
Osteogenesis Imperfecta
Achondroplasia
Specific Neural Tube Defects
Developmental Dysplasia of the Hip
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


35: Congenital Anomalies
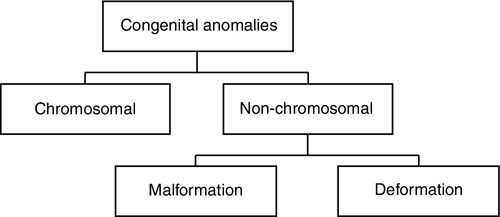
FIGURE 35-1 ■ Division of anomalies.
FIGURE 35-2 ■ Anterior view of dizygotic (fraternal) male twins discordant for Down syndrome (trisomy 21). The twin at right is smaller and hypotonic compared with the unaffected twin. The twin at right developed from a zygote that contained an extra 21 chromosome. (Courtesy Dr. A.E. Chudley, Department of Pediatrics and Child Health, University of Manitoba, Children’s Hospital, Winnipeg, Manitoba, Canada.)
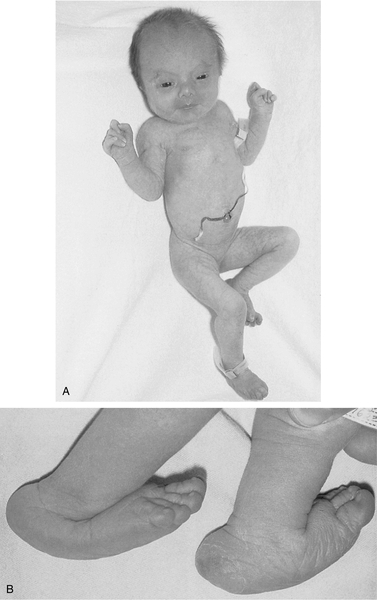
FIGURE 35-3 ■ A, Female neonate with trisomy 18. Note growth retardation, clenched fists with characteristic positioning of fingers (second and fifth digits overlap third and fourth digits). B, Feet of another trisomy 18 infant showing characteristic rocker-bottom appearance as a result of vertical position of tali (ankle bones). Also observe prominent calcanei (heel bones). (Courtesy Dr. A.E. Chudley, Department of Pediatrics and Child Health, University of Manitoba, Children’s Hospital, Winnipeg, Manitoba, Canada.)
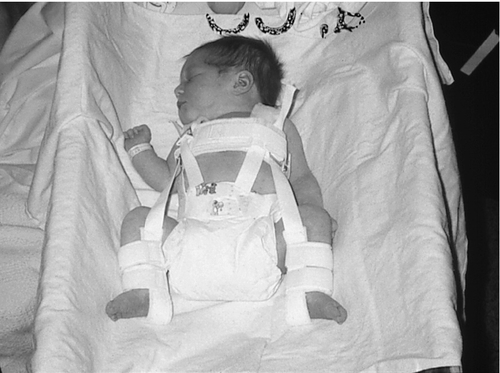
FIGURE 35-4 ■ Infant with dislocated hips in a Pavlik harness. (Courtesy Jane Deacon, RNC, MS, NNP, The Children’s Hospital, Denver, Colorado.)
