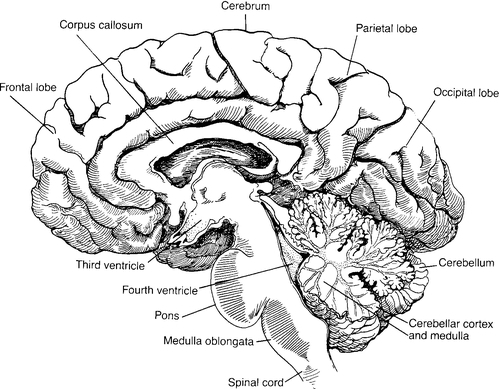
CHAPTER 34 2. Define autoregulation. 3. Review a complete neurologic examination. 4. Examine birth injuries and patient care management. 5. Differentiate between the different types of intracranial hemorrhages and their origins, clinical presentation, and outcomes. 6. Recognize neonatal seizures, their distinguishing characteristics, and issues in patient care management. 7. Describe hypoxic–ischemic encephalopathy. 8. Describe the clinical implications of periventricular leukomalacia. 9. Distinguish pathophysiologic factors, clinical presentation, and patient care management of early- and late-onset meningitis. The human brain is an intricate, fragile organ requiring precise development from the moment of conception. Several crucial developmental landmarks pinpoint major events in the development of the human brain. If the process is interrupted, difficulties ranging from simple, easily treatable conditions to major neurologic malformations may occur. Neurologic problems account for a significant number of admissions into the neonatal intensive care unit each year. This chapter provides a comprehensive review of neurodevelopment, neurophysiology, and neuromalformations. A. Embryologic development (Table 34-1). 1. Primary neurulation (dorsal induction). a. Occurs within the first month of life, ending between 24 and 28 days of gestation. b. Induction events of dorsal aspect of embryo. c. Neural tube is formed by the invagination and curling of the distal neural plate. d. Closure of the neural tube gives rise to the central nervous system (CNS), including the cranial nerves. e. This evolution results in the formation of the skull and vertebrae. f. Inaccuracies of primary neurulation result in craniorachischisis totalis, anencephaly, myeloschisis, encephalocele, myelomeningocele with Arnold–Chiari type II malformation. 2. Prosencephalic development. a. Peak development is in the second and third months of gestation. b. Inductive interactive events that occur primarily at the rostral end on the ventral aspect of the embryo. c. This influences the formation of the face, forebrain, corpus callosum and septum pellucidum, optic nerves/chiasm, the hypothalamic structures (thalamus and hypothalamus [diencephalon]), and the cerebral hemispheres (telencephalon). d. Absence of olfactory bulbs and tracts is not uncommon. e. Disturbance in prosencephalic development causes facial and forebrain alterations. (1) Holoprosencephaly (abnormal formation of telencephalon and diencephalon). (2) Midline and midfacial defects. (a) Hypotelorism (less common: hypertelorism). (b) Cyclopia. (c) Cleft lip with or without cleft palate. (3) Agenesis of corpus callosum, corpus pellucidum. f. Most common karyotype is normal (chromosomal disorder is possible). 3. Neuronal proliferation. a. Occurs initially at 2 months, with peak between 3 and 4 months of gestation. b. Toxins and inherited diseases can significantly alter the number of neurons. c. Chemical and environmental substances can reduce the number of neurons, causing microcephaly vera. d. Insufficient neurons in absence of apoptotic events are primary micrencephaly; excess neurons can produce macrencephaly. e. Disorders of proliferation of small veins cause Sturge–Weber syndrome (6% unilateral, 24% bilateral facial lesions). 4. Neuronal migration. a. Can occur as early as 2 months; peaks between 3 and 5 months. b. By 6 months of gestation, the neurons have migrated to their final, permanent place in the cortex. c. Neurons follow glial paths outward. d. Cells migrate and differentiate into six cortical layers. e. Migration is critical for development of the cerebral cortex and the deeper nuclear structures. (2) Hypothalamus. (3) Thalamus. (4) Brainstem. (5) Cerebellum. (6) Spinal cord. f. Dysfunction at this stage results in cortical malformation with abnormalities of neurologic function. g. Seizures may be the first clinical manifestation in the early postnatal period. h. Defects associated with abnormal migration range in severity and may be associated with other neurologic development. i. Abnormal development of gyrus denotes a neuronal migration disorder. j. Disorders include lissencephaly (“smooth brain”), pachygyria, agenesis of the corpus callosum, and schizencephaly (clefts found in the cerebral wall). 5. Neuronal organization. a. Peaks at 5 months of gestation to several years after birth. b. Provides the basis for brain function and its complex circuitry. c. Includes cell differentiation, cell death, synaptic development, neurotransmitters, and myelination. d. Achieves stabilization of cell connections. e. Disorders due to organizational deficits and detrimental retardation (as with Down syndrome, fragile X syndrome, and mild Duchenne’s muscular dystrophy). 6. Myelination. a. Begins in the second trimester and continues into adult life. b. Involves myelin deposition around axons. c. Myelin, a fatty covering, insulates the circuitry; prevents leakage of current and enables rapid, efficient transmission of nerve impulses. d. Enhances intercellular communication. e. Deficiencies occur in some acquired and inherited diseases. TABLE 34-1 Major Events in Human Brain Development and Peak Times of Occurrence From Volpe, J.J.: Neurology of the newborn (5th ed.). Philadelphia, 2008, Elsevier Saunders. B. Brain anatomy (Fig. 34-1) (Moore et al., 2012; Volpe, 2008). a. Promotes integrative muscle function. b. Maintains balance. c. Enables smooth, purposeful movements. 2. Cerebrum: main components of cerebral hemisphere. a. Contains four lobes: frontal, parietal, occipital, and temporal. (2) Parietal lobes are responsible for hearing, understanding speech, and forming an integrated sense of self. (3) Occipital lobes process vision. (4) Temporal lobes are centers for smell with associative areas for memory and learning. b. Corpus callosum: fiber bundles connecting the cerebral hemispheres. c. Cerebral cortex. (1) Encompasses the mind, the intellect. (2) Gray matter. d. Lateral ventricles. e. Third ventricle: fluid-filled space. f. Thalamus: integrates sensory input. g. Hypothalamus: regulates body temperature. 3. Brainstem. a. Relays input and output signals between higher brain centers and the spinal cord. b. Three main components. (a) Gives origin to cranial nerves VIII, IX, X, XI, and XII. (b) Controls areas of the abdomen, thorax, throat, and mouth. (2) Pons: carries information between the brainstem and the cerebellum. (3) Midbrain: involved in eye movements. 1. Cerebral metabolism is influenced by the availability of glucose and oxygen. 2. Glucose is transported from blood to brain by a glucose transporter found in capillaries. 3. Serum glucose provides the brain with a glucose pool. 4. The neonatal brain is glucose dependent. The CNS is quickly and significantly affected by hypoglycemia. 5. Glycogen stores are minimal or nonexistent in the premature baby. 6. The brain depends on adequate circulation to supply both oxygen and glucose to create enough energy for normal growth and metabolism. 7. Anaerobic metabolism causes lactic acid buildup. 8. Anaerobic metabolism produces significantly smaller amounts of energy. 9. Newborn blood glucose levels less than 30 mg/dL are associated with significant increases in cerebral blood flow. 10. Defining the lower limit of the neonatal blood glucose level is difficult because the infant’s ability to present overt symptoms of hypoglycemia is not developed. B. Cerebral blood flow. a. The brain increases cerebral blood flow to spare itself inadequacies. b. As pH decreases, cerebral blood flow increases. c. As potassium levels increase, cerebral blood flow increases. d. Hypoxemia causes an increase in cerebral blood flow to provide adequate oxygenated blood to the brain. e. Increased osmolarity causes increased cerebral blood flow. f. An increase in calcium ions causes a decrease in cerebral blood flow. g. Cerebral blood flow increases when blood glucose levels fall to less than 30 mg/dL; the hypoglycemic brain recruits previously unperfused capillaries to maintain glucose levels. Degree and duration of hypoglycemia are significant. h. Studies have shown that neonatal neurologic signs can be minimal or absent with subsequent abnormal cognitive development. 2. Autoregulation. a. Maintains steady-state cerebral blood flow over a broad range of perfusion pressures. b. Important vasoactive factors in the brain: hydrogen ions, potassium ions, adenosine, prostaglandins, osmolarity, calcium. c. Cerebral blood flow increases with advancing gestational age and concomitant cerebral metabolic demands. d. Normal arterial blood pressure in the preterm neonate is thought to be near or at the lower autoregulatory limit. This suggests an increased vulnerability to ischemic brain injury with modest hypotension, especially with decreasing gestational age. e. Cerebral vasculature vasodilates maximally in response to hypoxemia, hypercapnia, and acidosis. (1) Hypotension leads to ischemia. (a) Ischemia damages blood vessels and surrounding elements supporting the blood vessels. (b) Blood flow to cerebral white matter is restored only after reperfusion of other brain regions. (c) Once adequate blood supply resumes, hemorrhage can occur into ischemic areas. (2) Hypertension leads to hemorrhage. B. Observation. 1. Determine behavioral state. 2. Note posture. a. Gestational age determines posture. (1) Premature infants: open, extended position reflecting diminished tone. (2) Term infants: flexed position reflecting adequate tone. b. Sequelae of intrauterine position may be evident. c. Abnormal findings are as follows: (2) Asymmetry. (3) Flaccidity. 3. Note movements. a. Symmetrical or asymmetrical body movements. b. Note movement quality (jitteriness, seizures, tremors, and clonus). c. Quantity (absent or pronounced). 4. Note respiratory activity. b. Hypoventilation (apnea). c. Quality of cry. (1) High pitched (consider meningitis, drug withdrawal, neurologic abnormalities). (2) Stridor (consider vocal cord damage or paralysis). 5. Observe skin. a. Lesions (note number, size, shape, color, and texture). (1) Café-au-lait spots (six or more lesions of ≥ 1.5 cm; may indicate neurofibromatosis). (2) Port-wine facial hemangioma (consider Sturge–Weber syndrome). (3) Areas of depigmentation. b. Abrasions, lacerations, bruises, and forceps marks (consider intracranial bleeding, injury). C. Physical examination (Fenichel, 2007; Scher, 2013; Volpe, 2008). 1. Check the skull size, shape, symmetry, hair whorls, fontanelles, and sutures. 2. Measure frontal–occipital circumference (FOC). b. Document greater than 90th percentile (symmetrical vs. asymmetrical compared with total body growth; may indicate macrocephaly). 3. Examine the face for abnormalities in structure. b. Neck skinfolds. 4. Spine (intact, openings, masses). 5. Cranial nerve function. TABLE 34-2 Cranial Nerves Adapted from Scanlon, J.W., Nelson, T., Grylack, L.J., and Smith, Y.F.: A system of newborn physical examination. Baltimore, 1979, University Park Press; Hockenberry, M.J. and Wilson, D.: Wong’s nursing care of infants and children (9th ed.). St. Louis, 2011, Elsevier Mosby; Fletcher, M.A.: Physical diagnosis in neonatology. Philadelphia, 1997, Lippincott, Williams & Wilkins; http://library.med.utah.edu/pedineurologicexam/html/newborn_n.html#02; and http://www.fpnotebook.com/Nicu/Exam/NwbrnNrlgcExm.htm. EOM, Extraocular movements; PERL, pupils equal and reactive to light. b. May be difficult to assess in the preterm newborn. c. Blink reflex requires intact cranial nerves III and VII. d. Corneal reflex requires intact cranial nerves V and VII. (1) Generally elicited only if one suspects brain damage. (2) Consider testing integrity of reflex in the presence of eye damage. e. Cranial nerves IX, X, and XII regulate the tongue, swallow, gag, and cry. f. Rooting reflex starts at 28 weeks, with a complete response established by 32 weeks. g. Sucking reflex present at 28 to 30 weeks but slow, weak, and unsustainable; mature and robust by 34 to 36 weeks of gestation. h. Rooting and sucking reflexes test partial function of cranial nerves V, VII, and XII. i. Swallowing tests cranial nerves IX and X. 6. Muscle tone. a. Evaluate head lag, ventral suspension, clonus, and recoil from extension. b. Check symmetry; briskness versus flaccidity. 7. Reflexes. a. Check primitive reflexes (automatisms). (1) Blinking: An infant will close his or her eyes in response to bright lights. (2) Palmar and plantar grasp (bilaterally): An infant’s fingers or toes will curl around a finger placed in the area. (3) Babinski: As the infant’s foot is stroked, the toes will extend upward and fan outward. (4) Moro: A quick change in the infant’s position will cause the infant to throw back his or her head, extend his or her arms outward, and open the hands. (5) Startle: A loud noise will cause the infant to extend and flex the arms, while the hands remain in a fist. b. Evaluate symmetry and strength of response of all primitive reflexes. c. Consider clavicular or humeral fractures or brachial plexus injury in the presence of an abnormal Moro reflex. d. Grasp varies with gestational age; if grasp is absent, consider nerve damage. As the neural tube develops, developmental processes can be altered by intrinsic or extrinsic factors, resulting in defective closure or a reopening of the neural tube. Defects range from anencephaly to open or skin lesions of the spinal cord. A. Incidence and etiology. (Back and Plawner, 2012) 1. Neural tube defects are among the most commonly occurring congenital malformation of the CNS. a. Incidence of NTDs in the United States is estimated as 1 per 1000 live births (Moore et al., 2012). 2. Risks for recurrence are significantly higher if a previous pregnancy has resulted in a child with an NTD. The risk may actually be nearly triple for subsequent pregnancies. 3. The etiology of NTDs is multifactorial. This complex interaction of genetic and environmental factors operates independently to determine individual and population risk. The majority of defects occur sporadically. 4. The defects result from a failure in closure of the neural groove to form an intact neural tube. The type and severity of the defect is dependent on the location and extent of failure. 1. Anencephaly is the most common and severe of disorders of anterior neural tube closure, comprising over half of the NTDs (Back and Plawner, 2012). It is a primary defect in anterior neural tube closure. Presentation of the anomaly is variable, may extend from the level of the lamina terminalis to the foramen magnum. 2. Incidence is 1.0 to 10.0 per 1000 live births (Back and Plawner, 2012). Recurrence risk for parents who have had one affected child is 1.9% (Jones et al., 2013). 3. Genetic and environmental influences appear to be important in the development of the defect. B. Clinical presentation. 1. General: no documented growth deficiency. 2. Craniofacial: commonly involves the forebrain and upper brainstem, resulting in absence of the calvaria; cerebral hemispheres are missing. The lower brainstem is present, with rudimentary function. Hypothalamus and cerebellum are frequently malformed. Varied facial features, cleft palate and/or lip, altered auricular development, and cervical abnormalities are also common. (Back and Plawner, 2012). 3. Musculoskeletal: malformation of the ribs, thoracic cage, abdominal wall, anterior spina bifida, and short neck. C. Associated findings. 2. Diaphragmatic defects with or without herniation, hypoplastic lung. D. Diagnosis. 1. Fetal ultrasound or radiographs in the second trimester to detect absence of vital structures. 2. Maternal serum screening at 16 to 18 weeks identifies infants at risk. AFP is a major serum protein in the early embryo and is fetus specific. Leakage of fetal serum through an open NTD directly into the amniotic fluid results in elevation of AFP levels. These increases are detected by maternal serum and amniocentesis evaluation (Back and Plawner, 2012). E. Treatment/prevention. 1. U.S. Public Health Service recommends that women of childbearing age consume 0.4 mg of folic acid daily to decrease their risk for conceiving a child with an NTD. Periconceptional use of vitamins and folate have resulted in a 60% to 70% reduction in the recurrence of these malformations when given to women who had previously given birth to an infant with a neural tube disorder (Back and Plawner, 2012). In the United States, cereals and bread products are enriched with folate. 2. As most anencephalic infants are stillborn or die within a few days of delivery, the parents must prepare for the death of their infant at a time when they would be anticipating the birth. Support for the family must take into account their individual preferences and needs. 3. The opportunity for these infants to serve as organ donors is complex. Ethical and legal concerns over the diagnosis of brain death and persistent clinical signs of brainstem function have limited the donation of organs from anencephalic infants. Defects are secondary to restricted failure of the anterior neuropore to close at approximately day 26 of gestation, resulting in extension of the brain tissue through a defect in the skull. A. Incidence and etiology (Back and Plawner, 2012). 1. The defect occurs in 1 per 10,000 live births. In 70% to 80% of babies the lesion is found in the occipital region (Volpe, 2008). 2. Precise mechanism for development is unclear. 3. Genetic factors may be causative, autosomal recessive patterns of inheritance have been reported. Meckel’s syndrome and Walker–Warburg syndrome are the most commonly associated syndromes. 4. Environmental factors, including maternal teratogens, have been linked to development of encephalocele. a. Maternal febrile illness resulting in temperatures of 38.9° C (102.2° F) or greater within the first one third to one half of gestation have been found to be teratogenic (Jones et al., 2013). B. Clinical presentation. C. Associated findings (Back and Plawner, 2012). 1. Approximately 50% of infants presenting with an encephalocele have other major anomalies. Microcephaly, arrhinencephaly, cleft lip/palate, craniosynostosis, and hydrocephalus occur in approximately one half of the affected children. Agenesis of the corpus callosum occurs in two thirds of affected infants (Volpe, 2008). 2. Though normal intelligence is reported in approximately 50% of cases, intellectual deficiencies occur. 3. Hypotonia and associated motor defects. D. Diagnosis. 2. Chromosomal studies should be undertaken if suspicion of a genetic syndrome is present. 3. Careful history of maternal exposure to teratogens or adverse environmental events may provide clues to etiology. E. Treatment. 1. Based on the individual position and extent of the lesion. 2. For those families who experience a child with an encephalocele, genetic counseling is essential as recurrence risk is estimated at 3% to 5%, depending on the etiology of the defect. These spinal cord malformations include myelomeningocele and meningoceles and result from restricted failure of the posterior neuropore to close at approximately 26 days of gestation. A. Incidence and etiology (Volpe, 2008). 1. Overall incidence is 2 in 10,000 live births (Back and Plawner, 2012). 2. The etiology of these defects include: multifactorial inheritance, single mutant genes, chromosomal anomalies, and specific teratogens. a. Females are more commonly affected than males. b. The level of the myelomeningocele in the first affected infant has a direct effect on risk for subsequent infants. Recurrence risk for infants whose sibling had a lesion at T11 is 7.8%. If the index lesion was below T11, the subsequent risk was 0.7%. B. Clinical presentation (Volpe, 2008). 1. Defects are divided into two categories: (2) Approximately 80% of affected infants will exhibit a dermal lesion in the lumbosacral region. These defects include cutaneous dimples, cutaneous abnormalities or masses, and abnormal collections of hair. (3) Neurologic deficits are unusual in the newborn; sensory abnormalities in legs, feet or sphincter abnormalities may occasionally be present. b. Spina bifida manifesta: Spinal dysraphic conditions result from defects in caudal neural tube formation. (1) Meningocele: restricted herniation of meninges at the site of defect. Defect is uniformly localized to the lumbar region, is closed defect with intact dermal covering (Back and Plawner, 2012). (2) Myelomeningocele: characterized by herniation of meninges and spinal cord at the site of the defect. Lesions may or may not have vertebral or dermal covering. These defects are 4 times more common than meningoceles (Back and Plawner, 2012). (a) Rarely occurs as an isolated malformation, commonly associated with CNS abnormalities (Back and Plawner, 2012). C. Associated findings. 1. Hydrocephalus is a common associated development either at birth or shortly after. a. Development correlates with the site of the lesion: 60% in patients with occipital, cervical, or thoracic lesions; 90% in infants with thoracolumbar, lumbar, or lumbosacral lesions (Back and Plawner, 2012). 2. Infants with myelomeningocele experience serious urinary tract complications. These urinary issues are a major cause of death after the first year of life (Volpe, 2008). 3. Kyphosis at birth; scoliosis later in childhood. 4. Clubfeet secondary to neurologic and orthopedic abnormalities. 5. Dislocation of hips, delay in walking, asymmetry of legs. 6. Arnold–Chiari malformation occurs in 95% of patients with a lumbar lesion (Back and Plawner, 2012). Downward displacement of the medulla and the fourth ventricle through the foramen magnum into the upper cervical canal sets in motion a chain of events that are potentially lethal secondary to brainstem dysfunction and hydrocephalus. Development of a hoarse weak cry may indicate vocal cord paralysis and is a sign of herniation. 7. Tethered cord. 8. Seizure activity secondary to cortical dysgenesis occurs in 20% to 25% of infants with myelomeningocele (Volpe, 2008). D. Diagnosis. 1. AFP elevation in maternal serum at 16 to 18 weeks. 2. Amniotic fluid acetylcholinesterase and amniotic AFP at 14 to 16 weeks. 3. Fetal ultrasound. 4. Radiographs, CT, cranial ultrasonography, and MRI are done to provide visual record of anomalies. E. Treatment. 2. Clinical management is individualized to the type and position of the lesion. b. Assessment of function at the level of the lesion allows for estimates of potential capabilities; motor, sensory, and sphincter function. c. Examine lesion and measure size. d. Culture specimen from lesion if sac is open. Wrap lesion with sterile gauze moistened with warm sterile saline solution; place a sterile feeding tube within the gauze mesh for intermittent infusion of warm saline solution. Maintain the infant in a prone kneeling position, and protect the knees from skin breakdown. Place a drape over the buttocks below the lesion; utilize the drape’s adhesive backing to secure the drape to the body. e. Obtain immediate consultation: (2) Urology: evaluation must be undertaken to demonstrate deformity and determine the type and frequency of follow-up management needed. 3. Secondary to the extensive associated finding for these defects, parents must be made aware of the need for continuing care and support. Often a team approach is needed to ensure that complications are minimized. A. Microcephaly (Back and Plawner, 2012; Volpe, 2008). a. FOC ≥ 2 standard deviations below the mean for age and gender. b. Small brain implies neurologic impairment. 2. Risk factors. (2) Exposure to radiation. (3) Metabolic conditions such as diabetes or phenylketonuria. (4) Use of prescription and/or street drugs, especially in the first trimester. (5) Genetic foundation may be autosomal recessive, autosomal dominant, or X-linked. (6) Malnutrition is the most common etiology worldwide. b. Fetal: (1) Prenatal/perinatal insult: inflammation; hypoxia; birth trauma. c. Neonatal: (1) Very low birth weight infant. (2) Hypoxic–ischemic encephalopathy (HIE). (3) Nutrition: most common worldwide cause. 3. Pathophysiology. a. Neuronal proliferation defect. b. Occurs between 3 and 4 months of gestational age. c. Destructive microcephaly occurs when the normal brain suffers prenatal/perinatal insult. 4. Clinical presentation. a. Small head, backward sloping of the forehead, small cranial volume. b. Neurologic deficits rarely evident at birth. 5. Diagnostic evaluation. a. Perform a complete physical examination, including neurologic assessment. b. Elicit a thorough maternal history. c. Use tests to confirm or rule out etiologic factors aligned with maternal history. d. Computed tomography (CT) or magnetic resonance imaging (MRI) is performed. 6. Patient care management. a. Record accurate measurement of FOC, length, and weight weekly. b. Note percentiles and alert physician to abnormalities. c. Document clearly any deviations from normal. d. Obtain tests as ordered; note dates to follow up results. e. Ensure that the family is informed. f. Obtain consultations as needed: genetics, infectious diseases. 7. Outcome. b. May be associated with developmental delays. B. Hydrocephalus (Prabhu et al., 2012; Verklan and Lopez, 2011; Volpe, 2008). a. CSF is in balance between formation and absorption. b. CSF is produced at a rate of 0.35 mL/min from brain parenchyma, cerebral ventricles, areas along the spinal cord, and the choroid plexus (70% is from the choroid plexus). 2. Pathophysiology. a. Excessive CSF production (rare). b. Inadequate CSF absorption secondary to abnormal circulation. c. Excess ventricular CSF secondary to aqueductal outflow obstruction causes obstructive, noncommunicating hydrocephalus (refer to Fig. 34-2 for a simplified diagram of the brain). (1) The condition is most common in newborn infants. (2) Obstructive hydrocephalus may progress rapidly. d. Excess ventricular CSF with flow between the lateral ventricles and the subarachnoid space results in communicating, nonobstructive hydrocephalus. 3. Congenital hydrocephalus. (2) Dandy–Walker cyst (cystic transformation of fourth ventricle). (3) Myelomeningocele with Arnold–Chiari malformation (herniation of the hindbrain, usually causing obstructive hydrocephalus). (4) Congenital masses and tumors. (5) Congenital infection (toxoplasmosis, cytomegalovirus infection). b. Associated etiologies and/or congenital defects. (2) Encephalocele. (3) Holoprosencephaly. c. Clinical presentation. (2) Widened sutures. (3) Full (bulging) and tense fontanelles. (4) Increasing FOC. (5) “Setting-sun” eyes (may signify brain tissue damage). (6) Vomiting, lethargy, irritability. (7) Visible scalp veins. d. Diagnostic evaluation. (1) Serial intracranial ultrasonography. (2) Neuroimaging techniques: CT, MRI, and cranial ultrasonography. e. Patient care management. (2) Perform a thorough physical examination, assessing for further anomalies. (3) Obtain neurosurgery and genetics consultations. (4) Confirm diagnosis and cause. (5) Consider the possible need for reservoir placement versus ventriculoperitoneal (VP) shunt placement. (6) Support the infant by decreasing noxious stimuli (dim lights, minimal handling). (7) Position the head carefully. (8) Provide normal infant care as much as possible. (9) Involve parents in infant’s care as soon as family is ready. (10) Allow parents to view an infant with a VP shunt or review pictured handouts. (11) Review VP shunt with parents preoperatively and postoperatively. (12) Prevent skin breakdown by not allowing the infant to put his or her head on the shunt side postoperatively. (13) Relieve the infant’s probable stiff neck by holding the child’s neck on the shunt side during feedings. (14) Review signs of infection or blocked shunt with the family. (b) Vomiting. (c) Increasing head size. (d) Lethargy. (e) Changes in feeding patterns. (f) Bulging fontanelle. (15) If incision site reddens, position infant on opposite side to relieve pressure from this area. 4. Posthemorrhagic hydrocephalus (Prabhu et al., 2012; Volpe, 2008). (2) Two types: acute and chronic. (i) Rapidly appears—within days of the initial hemorrhage. (ii) Probably occurs secondary to malabsorption of CSF secondary to a blood clot. (b) Subacute, chronic. (ii) Blood from IVH. b. Incidence. (1) Approximately 45% of infants with IVH have no evidence of hydrocephalus (Volpe, 2008). (2) Acute ventricular dilatation develops in approximately 50% of surviving infants with hemorrhage; in the majority, it resolves spontaneously or remains static (Papile, 2006). c. Clinical presentation. (1) Insidious following mild ventricular dilatation. (2) May be profound following severe ventricular dilatation. (a) Rapid increase in head size (begins days to weeks after ventricular dilatation present). (b) Episodic apnea and bradycardia. (c) Lethargy. (d) Increased intracranial pressure. (e) Tense, bulging anterior fontanelle. (f) Cranial sutures separating. (g) Ocular movement abnormalities. d. Diagnostic evaluation. (1) Graph of weekly FOC measurements. (2) CT scan. (3) Cranial ultrasonography. (4) MRI. e. Patient care management (Verklan, 2012; Verklan and Lopez, 2011; Volpe, 2008). (1) Obtain daily FOC measurements. (2) Serial cranial ultrasonography. (3) Neurosurgical consultation. (4) Interventions to maintain lumbar or ventricular pressure at approximately 5 cm H2O while evaluating for shunt placement. (a) Serial lumbar punctures or direct ventricular access may be helpful. (b) Administer medications that diminish CSF production rates: (i) Furosemide (Lasix): 1 mg/kg/day. (ii) Acetazolamide (Diamox): up to 100 mg/kg/day. (5) Consideration given to placing a reservoir or VP shunt. (6) Observe the infant for signs of increasing intracranial hemorrhage and hydrocephalus. (7) Support the family: neonate is very susceptible to shunt infections and shunt malfunction. f. Outcome (Prabhu et al., 2012; Verklan, 2012; Volpe, 2008). (1) Poor outcomes are likely when cerebral decompression does not occur after VP shunt placement. (2) Initial IVH severity is the major determining factor in posthemorrhagic hydrocephalus development. (3) In slightly more than 50% of the cases, severe hemorrhage results in progressive ventricular dilatation. (4) Without therapy, a considerable number of infants exhibit halted progression, with or without resolution. (5) Deficits are motor and/or cognitive. C. Craniosynostosis (Evans et al., 2012). a. Premature closure of cranial sutures. b. Occurs along one or more suture lines (see Fig. 34-3 for names and placement of cranial sutures).
Neurologic Disorders
ANATOMY OF THE NEUROLOGIC SYSTEM (MOORE ET AL. 2012; VOLPE, 2008)
Major Developmental Event
Peak Time of Occurrence
Primary neurulation
3 to 4 weeks of gestation
Prosencephalic development
2 to 3 months of gestation
Neuronal proliferation
3 to 4 months of gestation
Neuronal migration
3 to 5 months of gestation
Organization
5 months of gestation to years postnatal
Myelination
Birth to years postnatal
PHYSIOLOGY OF THE NEUROLOGIC SYSTEM (McLEAN ET AL., 2012; VOLPE, 2008; YAGER, 2012)
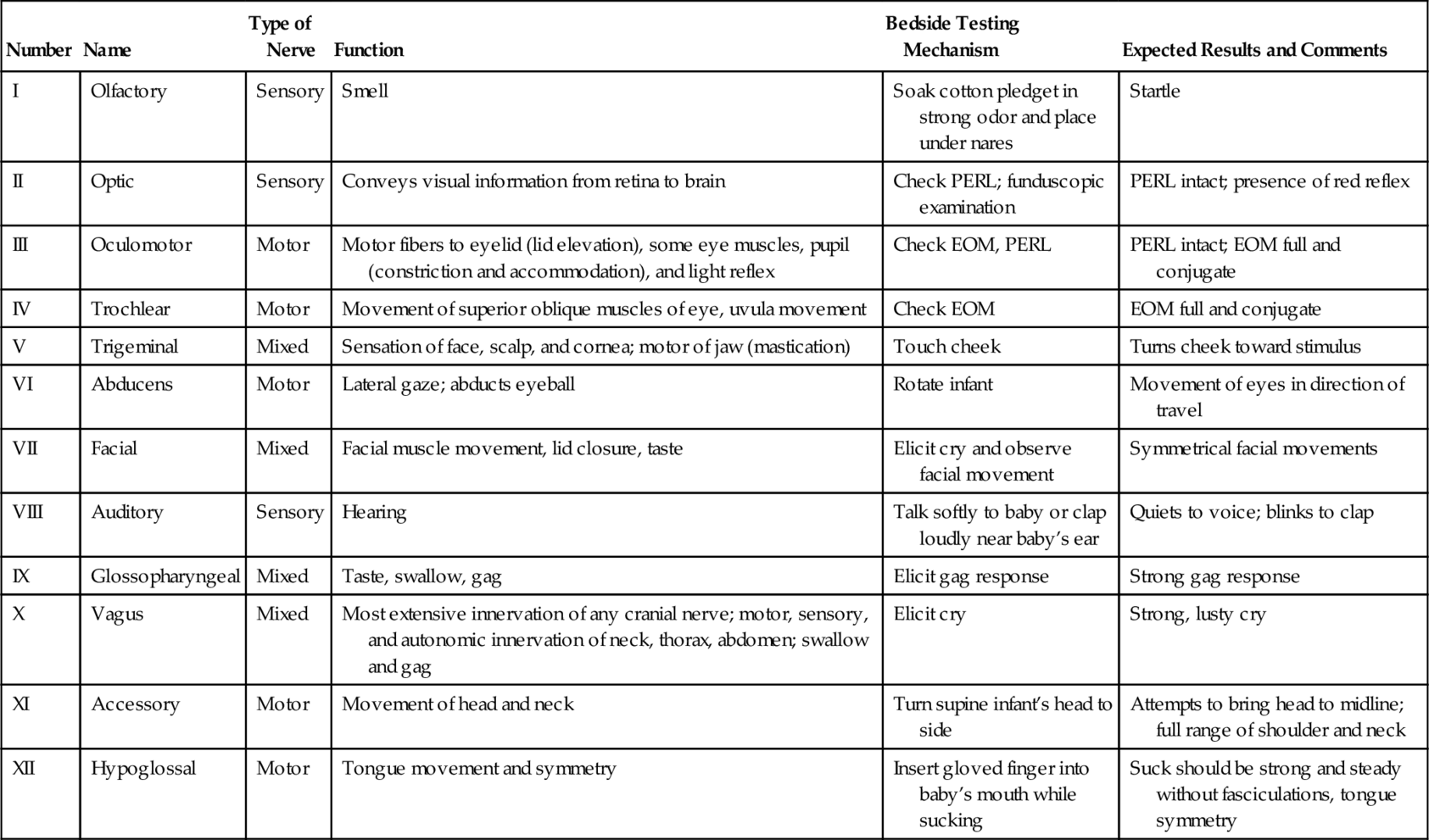
NEUROLOGIC ASSESSMENT (RENNIE AND HUERTAS-CEBALLOS, 2012; VOLPE, 2008)
Number
Name
Type of Nerve
Function
Bedside Testing Mechanism
Expected Results and Comments
I
Olfactory
Sensory
Smell
Soak cotton pledget in strong odor and place under nares
Startle
II
Optic
Sensory
Conveys visual information from retina to brain
Check PERL; funduscopic examination
PERL intact; presence of red reflex
III
Oculomotor
Motor
Motor fibers to eyelid (lid elevation), some eye muscles, pupil (constriction and accommodation), and light reflex
Check EOM, PERL
PERL intact; EOM full and conjugate
IV
Trochlear
Motor
Movement of superior oblique muscles of eye, uvula movement
Check EOM
EOM full and conjugate
V
Trigeminal
Mixed
Sensation of face, scalp, and cornea; motor of jaw (mastication)
Touch cheek
Turns cheek toward stimulus
VI
Abducens
Motor
Lateral gaze; abducts eyeball
Rotate infant
Movement of eyes in direction of travel
VII
Facial
Mixed
Facial muscle movement, lid closure, taste
Elicit cry and observe facial movement
Symmetrical facial movements
VIII
Auditory
Sensory
Hearing
Talk softly to baby or clap loudly near baby’s ear
Quiets to voice; blinks to clap
IX
Glossopharyngeal
Mixed
Taste, swallow, gag
Elicit gag response
Strong gag response
X
Vagus
Mixed
Most extensive innervation of any cranial nerve; motor, sensory, and autonomic innervation of neck, thorax, abdomen; swallow and gag
Elicit cry
Strong, lusty cry
XI
Accessory
Motor
Movement of head and neck
Turn supine infant’s head to side
Attempts to bring head to midline; full range of shoulder and neck
XII
Hypoglossal
Motor
Tongue movement and symmetry
Insert gloved finger into baby’s mouth while sucking
Suck should be strong and steady without fasciculations, tongue symmetry
NEURAL TUBE DEFECTS (NTDs)
Anencephaly (Volpe, 2008)
Encephalocele
Spina Bifida
NEUROLOGIC DISORDERS
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


34: Neurologic Disorders
FIGURE 34-1 ■ Anatomy of the brain.
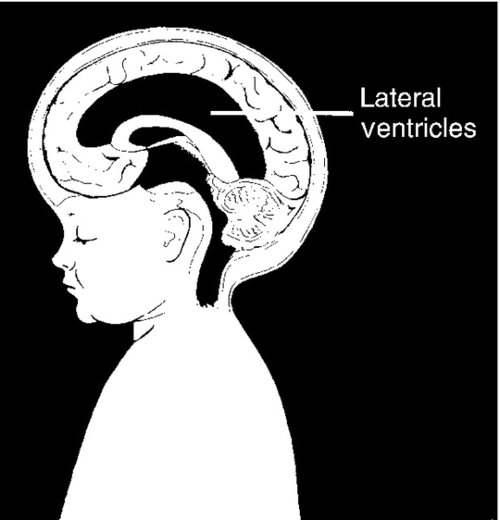
FIGURE 34-2 ■ Hydrocephalus. (From Ross Laboratories: New perspectives on intraventricular hemorrhage. Columbus, OH, 1988, Ross Laboratories.)
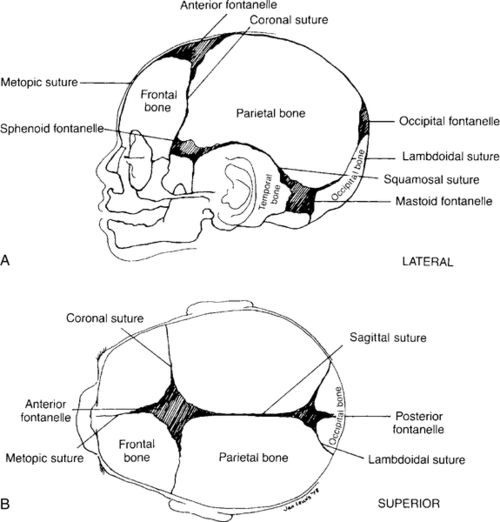
FIGURE 34-3 ■ A and B, Two views of neonatal skull, showing clinically important fontanelles and sutures. (From Scanlon, J.W., Nelson, T., Grylack, L., and Smith, Y.F.: A system of newborn physical examinations. Baltimore, 1979, University Park Press, p. 47.)
