CHAPTER 30 1. Define the endocrine system. 2. Describe endocrine system regulation. 3. Identify and discuss endocrine disorders that manifest in the neonatal period, including disorders of the thyroid, pituitary, adrenal gland, pancreas, and genital development. 4. List effective ways to help parents cope with the birth of an infant with a disorder of sexual development. Disorders of the endocrine system are relatively rare in neonates, but usually have lifelong consequences. The classic endocrine system is a group of nine ductless glands (Table 30-1), but in reality it includes every organ and cell in the body that produces and responds to hormones. A system of communication between different cells of the body, the endocrine system is intricately linked with the neurologic and immune systems in a vast, interacting control network (Wilson, 2005). Coordination and regulation of metabolism and energy, the internal environment, growth and development, and sexual differentiation and reproduction are among the functions of this complex system. TABLE 30-1 Major Glands and Hormones of the Endocrine System 2. Hormones bind to specific receptors on the surface of or within the cytoplasm or nucleus of target cells to produce physiologic actions. Sensitivity of a target cell to its hormones is critical to normal function. 3. Many hormones are secreted directly into the circulation for transport to various target tissues. Hormones can also act on cells in the immediate vicinity of their release (paracrine action) or on the cell that produced the hormone (autocrine or intracrine action). 4. Some hormones circulate partly in free form and partly bound to transport proteins. It is the free form that is available for receptor binding and that dictates regulatory influences on hormone release. Clinical states of hormone excess and deficiency correlate best with free hormone levels. 5. Most hormones are secreted in their biologically active form (such as insulin), but others (such as thyroxine) must be converted to their final active form in peripheral tissues. B. Endocrine system regulation (Fig. 30-1). 2. Other endocrine glands, such as the parathyroids and the pancreatic islets, are not part of the hypothalamic–pituitary axis but have “freestanding” control mechanisms. These glands release hormones that stimulate a target tissue to produce an effect, which in turn directly modifies the output of the gland. 3. The neuroendocrine system plays a key role in body homeostasis. Hormones act as neurotransmitters, and neurotransmitters are involved in regulating endocrine function. Endocrine glands such as the hypothalamus, the pituitary, and the adrenal cortex respond to neural stimulation. The neuroendocrine system is important in the smooth adaptation of the neonate to the stresses of extrauterine life. C. Endocrine disruptors. 2. Exposure to an endocrine disruptor during embryonic gonadal sex differentiation can alter male germline epigenetics, a mechanism that can transmit adult-onset diseases, such as spermatogenic defects, prostate disease, kidney disease, and cancer, to both current and future generations (Anway and Skinner, 2008). 3. Sources of these chemicals include pharmaceuticals, dioxin and dioxin-like compounds, polychlorinated biphenyls, dichlorodiphenyltrichloroethane (DDT) and other pesticides, and plasticizers such as bisphenol A. D. Fetal origins of adult disease. 2. Chronic stress may induce the “thrifty phenotype fetus” and permanently alter the fetal hypothalamic–pituitary axis. In response to stressors, the fetus generates glucocorticoids and catecholamines that program the fetal endocrine system. Fetal endocrine programming has been associated with endocrine, metabolic, and cardiovascular disease in the adult. E. Endocrine disorders in the neonate. 2. Anterior pituitary arises from the oral ectoderm and its cells to differentiate into specific hormone-producing cells. The release of trophic hormones—growth hormone (GH), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), thyroid-stimulating hormone (TSH), luteinizing hormone (LH), and prolactin (PRL)—is influenced by hypothalamic releasing/inhibiting hormones. 3. Posterior pituitary develops from neuroectoderm evaginating ventrally from the brain. The posterior pituitary produces oxytocin and antidiuretic hormone (ADH). B. Hypopituitarism. 1. Although rare in the newborn, hypopituitarism can be congenital or acquired by infection, hypovolemic shock, precipitous delivery, or low Apgar scores. Congenital hypopituitarism can be caused by mutations in signaling molecules of transcription factors regulating pituitary gland development, or may be part of a syndrome such as optic nerve hypoplasia, and be associated with absent septum pellucidum (Garcia-Fillon and Borchert, 2013). Congenital hypopituitarism can overlap with hypogonadotropic hypogonadal disorders such as Kallmann syndrome (Bancalari et al., 2012). 2. Types of hypopituitarism include absence of pituitary gland (pituitary agenesis), panhypopituitarism (deficiency of all pituitary hormones), and an isolated hormone defect, such as GH deficiency. A primary hypothalamic disorder can also result in hypopituitarism. 3. Neonates with congenital hypopituitarism can initially be asymptomatic, with evidence of pituitary hormone deficiencies developing over time (Alatzoglou and Dattani, 2009). Neonates with hypopituitary syndromes can present with midline craniofacial defects such as cleft lip, cleft palate, bifid uvula, or micropenis in boys (a normally formed and proportioned penis with a stretched penile length more than 2 standard deviations below the mean for age) (Tuladhar et al., 1998). Hypoglycemia can be mild or severe and persistent. 4. Later in the neonatal period, infants may present with prolonged jaundice and direct hyperbilirubinemia, or evidence of other endocrinopathy, such as diabetes insipidus (high urine output, dehydration, hypernatremia). 5. Diagnosis is made by hormone testing (including GH, thyroid hormone, cortisol), ultrasound or magnetic resonance imaging (MRI) of anatomic structures, and genetic analyses. Tests of pituitary function include stimulation tests (ACTH, thyrotropin-releasing hormone [TRH], glucagon) to test the response of pituitary hormones. A pediatric endocrinologist usually coordinates the diagnostic testing and interpretation for these infants. 6. Management involves correcting hypoglycemia and replacing deficient hormones. C. Diabetes insipidus. 2. Up to 90% of neonates with inherited nephrogenic DI are boys with an X-linked form caused by mutations in the arginine vasopressin receptor 2 gene (AVP2R) (Copelovitch and Kaplan, 2012). 3. ADH secretion is normally triggered by changes in osmolality, increasing or decreasing to regulate urine output. In DI, ADH is deficient, resulting in free water loss. 4. Clinical manifestations are vigorous sucking followed by vomiting, polyuria (high urine output with low specific gravity), irritability, fever, and evidence of dehydration. 5. Diagnosis is made by reviewing serum electrolytes, osmolality, plasma ADH levels, genetic analysis, urine output, and urinalysis. MRI may be done to visualize the posterior pituitary. 6. DI is treated with cautious fluid management to correct dehydration without causing rapid shifts in serum sodium levels. Desmopressin (DDAVP) may be required to supplement ADH if the neonate is unable to concentrate urine. D. Syndrome of inappropriate ADH. 1. Impairment of free water clearance caused by an uncontrolled release of ADH. 2. Causes include CNS infection, birth asphyxia, intracranial hemorrhage, and meningitis. The release of ADH is inappropriate to the level of osmolality in the serum, causing fluid retention, oliguria, hyponatremia, weight gain, and edema. 3. Diagnosis is made by finding an elevated circulating ADH level with low serum osmolality and hyponatremia. 4. Treatment involves fluid restriction and monitoring of electrolytes, glucose, intake, and output. If it is not possible to manage DI with fluids alone, the agent of choice for pharmacologic treatment is DDAVP, a long-acting synthetic analogue of pituitary ADH. B. Normal physiology. 1. Functions of the thyroid gland. a. Concentrates and stores iodide, a trace element required for thyroid hormone synthesis. b. Synthesizes thyroglobulin, a thyroid hormone precursor. c. Synthesizes and releases the thyroid hormones thyroxine (T4) and triiodothyronine (T3), catalyzed by the enzyme thyroid peroxidase (TPO). 2. Thyroid hormone metabolism. b. T4 enters the cell and is converted to T3 by enzymes. c. Deiodination of the outer ring of T4 produces T3. Deiodination of the inner ring of T4 produces reverse T3 (rT3), a biologically inactive product. 3. Thyroid hormone transport. b. TBG, synthesized in the liver, has a high affinity for T3 and T4, carrying 70% of circulating hormone. When TBG is deficient, total thyroid hormone concentrations may be lower but free hormone levels are normal. 4. Mechanisms of thyroid gland regulation. b. Deiodinase enzymes in the anterior pituitary, brain, heart, liver, and other tissues regulate intracellular T3 availability. c. Autoregulation of hormone synthesis by the thyroid gland itself in relationship to its iodine supply. d. Stimulation or inhibition of thyroid function by TSH receptor antibodies. 5. Physiologic effects of thyroid hormones. b. Growth and differentiation of organs and tissues, including the bones, lungs, and CNS. Thyroid hormones induce differentiation and maturation of neural circuits during critical periods of brain development. Absence of thyroid hormones delays critical events by interrupting intercellular communication (Brook and Dattani, 2012). C. Fetal thyroid development. 1. The thyroid is the first endocrine gland to develop in the fetus, originating at 3 to 4 weeks of gestation (Forghani and Aye, 2008). 2. At about 10 to 12 weeks of gestation, the hypothalamus begins synthesizing TRH, the pituitary gland begins secreting TSH, and TBG is detectable in fetal serum. 3. The HPT axis begins to function at midgestation (weeks 18 to 20), when iodide uptake increases and the fetal thyroid gland begins to release T4. Total and free T4, TSH, and TBG increase steadily until term. The T3 level remains low until 30 weeks of gestation, rising only in the last 10 weeks as mechanisms for deiodination of T4 in fetal tissues mature. 4. Before 20 weeks of gestation, transplacental passage of maternal T4 largely provides for fetal thyroidal needs, and is critical for normal development. Maternal hypothyroidism during early gestation can impair CNS development in the fetus. By the start of the second trimester, however, the fetal contribution to circulating thyroid hormones is significant. 5. The fetus is also dependent on the maternal–placental system for adequate supply of iodide, a critical substrate for fetal thyroid hormone synthesis. Autoregulation of iodide uptake is not yet mature, so the fetal thyroid is susceptible to inhibitory effects of both iodide deficiency and iodide excess. 6. Cord blood TSH and T4 are directly proportional to birth weight and gestational age. At birth, 30% of thyroid hormone circulating in the infant is of maternal origin (Forghani and Aye, 2008). D. Neonatal thyroid physiology. 2. The TSH surge stimulates an abrupt rise in thyroid hormone levels. T4 and T3 both increase in response to TSH, peaking at 24 to 36 hours after birth. This physiologic hyperthyroid state is temporary, and occurs in response to sudden exposure to a cold environment. T4 increases in most infants to a level of 6.5 mcg/dL or higher. The rise in T4 causes the TSH to decline to 20 mU/l or less (the cutoff used in most screening programs for congenital hypothyroidism) because of feedback inhibition. 3. Postnatal changes in TSH, T4, and T3 occur in less mature infants as well but are quantitatively lower. The extremely preterm infant exhibits a dramatic fall in T4 over the first 1 to 2 weeks of life. Hypothyroidism in the neonate can be considered either permanent (lifelong therapy required) or transient (spontaneously resolving in weeks or months; treatment is temporary or not required at all). Hypothyroidism can also be termed congenital (existing at birth) or acquired. Other labels indicate the origin of the hypothyroidism: 2. Central (also called secondary/tertiary). Deficient thyroid hormone secretion due to a disorder affecting pituitary control (TSH production) or hypothalamic control (TRH production). A. Etiologies of permanent congenital hypothyroidism (CH). a. Thyroid dysgenesis: absent (agenesis/athyreosis), hypoplastic, and/or ectopic (usually sublingual) gland. Ectopic gland is the most common etiology of CH. Familial thyroid dysgenesis is primarily caused by mutations in the thyrotropin receptor gene TSHR (Cangul et al., 2012). b. Familial dyshormonogenesis: inborn errors of thyroid hormone biosynthesis, transport, or metabolism. About 10% to 20% of infants with CH have inherited defects of thyroid hormone metabolism, the most common of which is an organification defect related to deficient activity of the TPO enzyme. 2. Extrathyroid abnormalities. a. Defects of the pituitary gland (e.g., hypopituitarism) or the hypothalamus. b. TBG deficiency: X-linked disorder; more common in males. c. Thyroid hormone resistance. Resistance to the actions of endogenous and exogenous T4 and T3, a form of “peripheral hypothyroidism,” is becoming more common. All serum thyroid hormone levels are elevated, and many patients have goiter. B. Etiologies of transient hypothyroid states. b. Drugs given to the mother that cross the placenta and affect fetal thyroid production (propylthiouracil, methimazole, lithium, phenytoin, amiodarone, and radioiodine.) c. Ingestion of excess iodine (Connelly et al., 2012) or severe dietary iodine deficiency (Zimmermann, 2012). 2. Postnatally acquired: transiently impaired thyroid hormone production from exposure to iodine-containing topical disinfectants, ointments, drugs such as amiodarone, or intravenously administered contrast media. Preterm infants can absorb and excrete large amounts of iodine; thus, exposure to iodinated products should be minimized. C. The preterm infant: In addition to the same incidence of permanent CH as full term infants, several transient hypothyroid states have been described in preterm infants. Because two or more transient conditions can coexist, it is not always possible to determine the precise cause of low thyroid hormone levels in preterm infants. 1. Hypothyroxinemia of prematurity. Serum levels of thyroid hormones in preterm neonates are significantly lower and more variable than those of term neonates and correlate with gestational age and birth weight (Fisher, 2007). T4 levels of most preterms reach a nadir at 7 to 14 days of age and then climb to normal within 4 to 8 weeks. 2. Sicker preterm infants demonstrate more variability in their T4 values than healthy preterms of the same gestational age. Preterm infants do have an initial TSH surge after birth, but it is blunted in comparison to more mature neonates (Clemente et al., 2007). The TSH of preterm infants with hypothyroxinemia is not consistently elevated, suggesting relative lack of hypothalamic response to the lower T4 level. This condition is believed to be a developmental phenomenon caused by one or more of the following: a. Immaturity of thyroid hormone metabolism and the HPT axis. b. Loss of maternal contributions to the thyroid hormone pool at birth. c. Low TBG levels. d. Increased use of T4 to meet the demands of extrauterine life. e. Insufficient enteral or parenteral iodine intake. 3. Nonthyroidal illness syndrome. In some ill preterm infants, T4 is preferentially converted to rT3 instead of T3, possibly as an adaptive response to lower the metabolic rate during times of severe illness. The outcome is low serum concentrations of both T4 and T3. rT3 is elevated and TSH is normal. This condition, also known as “low T3 syndrome” or “euthyroid sick syndrome,” occurs in infants who have immature lungs or infections, because the cytokines produced in response to illness or inflammation are believed to inhibit thyroid function, metabolism, or action (van Wassenaer and Kok, 2004). The low T4 from nonthyroidal illness reverses spontaneously when the infant’s condition improves without treatment. Similar effects are seen in infants who are receiving dopamine or glucocorticoids, both of which can lower serum T4 concentrations. D. Clinical presentation and assessment. 2. The signs and symptoms of hypothyroidism in the neonate reflect the wide-ranging actions of thyroid hormones on metabolism, intestinal motility, cardiac function, temperature regulation, neurologic function, and skeletal maturation (Box 30-1). The possibility of CH must be considered in infants presenting with birth weight in excess of the 90th percentile, prolonged jaundice, hypothermia and cold mottled skin, an enlarged (> 1 cm) posterior fontanelle, umbilical hernia, and failure to feed well (Rastogi and LaFranchi, 2010). 3. Other features traditionally associated with hypothyroidism (macroglossia, dry skin, lethargy, hypotonia, hoarse cry, coarse hair, and constipation) evolve over the first weeks of life. A palpable, enlarged thyroid gland (also called a goiter) can be associated with impaired thyroid hormone synthesis and hypothyroidism. Hyperplasia of the thyroid gland results from hypersecretion of TSH in response to low T3 and T4 levels. Infants with suspected central hypothyroidism may present with midline or cranial defects or other signs of pituitary deficiency. Central hypothyroidism should be suspected in infants presenting with optic nerve hypoplasia, hypoglycemia, micropenis, or cleft lip/palate. E. Diagnostic studies in hypothyroidism (Table 30-2): Unless the infant is born to a mother with a history of thyroid dysfunction or has obvious clinical signs of hypothyroidism at birth, the diagnosis is usually made after the infant is identified by neonatal screening. 1. Newborn screening for hypothyroidism. b. Owing to the physiologic surge in TSH in the first hours of life, the specimen must be collected when the infant is at least 24 hours of age, and preferably between 2 and 4 days of age. If collected earlier, particularly in the first 3 hours of life, a false-positive result can occur. In those instances, a repeat specimen must be collected within the first 7 days of life, regardless of first test results. c. The incidence of CH, as detected through newborn screening, is approximately 1 per 3000 to 4000. An elevated TSH level (> 40 mU/l) is presumed to be CH until further testing proves otherwise. Rapid confirmation is essential. d. Screening errors caused by improper specimen collection, storage, or transport can result in a false-negative test. Thyroid medications taken by the mother during pregnancy can also produce false-negative results. e. Approximately 10% of infants with CH are missed on initial screening; they are detected only through routine second screening in states where this is required or on clinical grounds. Some of these infants have compensated hypothyroidism or delayed rise in the TSH level; most seem to have milder forms of hypothyroidism but still require treatment. f. Infants at risk of a missed or delayed diagnosis are those born at home, those who were extremely ill in the neonatal period, and those who were transferred to another hospital at an early age. 2. Thyroid function tests. a. A serum T4 less than 6 mcg/dL and a serum TSH greater than 50 mU/L strongly suggest CH. A normal T4 (e.g., > 10 mcg/dL) with elevated TSH suggests enough functional thyroid tissue to respond to excess TSH stimulation (seen in compensated or subclinical hypothyroidism). Free T4 and T3 levels may also be measured, along with other tests, as needed, to determine the cause of abnormal screening results (see Table 30-2). b. Age-related reference norms, for both gestational age and hours of age, should be used when interpreting all thyroid test results. c. If maternal antibody-mediated hypothyroidism is suspected, maternal antithyroid antibody (TRBAb) testing is done. Other thyroid autoantibodies that can produce hypothyroidism include thyroglobulin antibodies (TGAb) and thyroid peroxidase antibodies (TPOAb). d. TBG levels can be measured to rule out TBG deficiency. Thyroglobulin levels in infants with possible CH can help to differentiate between thyroid agenesis and dyshormonogenesis, as an adjunct to thyroid imaging. 3. Imaging studies: Further evaluation for the cause of a CH blood profile includes Doppler ultrasonography of the neck, thyroid radionuclide imaging (to identify normal or ectopic thyroid tissue), and bone age radiography of the knee or foot (delayed bone ossification suggests long-standing thyroid deprivation). TABLE 30-2 Summary of Low Thyroid States in the Newborn Infant T3, Triiodothyronine; T4, thyroxine; TBA, thyroid-blocking antibodies; TBG, thyroid-binding globulin; TFTs, thyroid function tests; TSH, thyroid-stimulating hormone; TSI, thyroid-stimulating immunoglobulins; VLBW, very low birth weight. * T4 low: less than 6 mcg/dL. † TSH elevated: greater than 40 mU/l; TSH normal: less than 10 mU/l; TSH borderline: 20 to 40 mU/l. Note that a slightly elevated TSH level may be normal in the first 2 days of life. ‡ TFTs: May include assays of total and free T4 and T3 along with TSH. § Tg: Thyroglobulin, a thyroid hormone precursor produced by the thyroid gland. A low level suggests thyroid agenesis. {ParaMarks} T3, resin uptake level: an indirect measure of protein binding. A high level suggests low binding capacity, as in TBG deficiency. ¶ TRH stimulation is a test of hypothalamic or pituitary control of thyroid function. A dose of thyrotropin-releasing hormone (TRH) is administered and TSH is measured serially. A subnormal TSH response suggests a deficient pituitary gland, and a delayed response suggests hypothalamic congenital hypothyroidism. F. Management. a. Early, adequate treatment of CH is critical for optimal neurologic development. The goal of hormone replacement therapy is to rapidly normalize the infant’s serum T4 level, and maintain it in the upper half of the normal range, which should result in a TSH of 0.5 to 4 mU/l (Bollepalli and Rose, 2012). The agent of choice is sodium levothyroxine because it is substantially converted to T3 within the brain. b. Infants receiving thyroid replacement therapy must be monitored closely for adequacy of treatment and evidence of thyrotoxicosis (irritability, tachycardia, poor weight gain). Serum T4 should normalize in 1 to 2 weeks; serum TSH can take longer to normalize. (2) Undertreatment leads to clinical hypothyroidism, delayed bone maturation, and neurologic damage. 2. Transient hypothyroidism. a. Hypothyroxinemia of prematurity is associated with higher mortality and neurodevelopmental deficits, yet cumulative evidence to date has not been able to demonstrate clear benefits of routinely supplementing these infants with thyroxine during early life (La Gamma and Paneth, 2012). A Cochrane Database review did not support the use of prophylactic thyroid hormones in preterm infants to reduce neonatal mortality or morbidity or improve neurodevelopmental outcomes (Osborn and Hunt, 2007). The exception is the infant with an elevated TSH level; these infants require treatment. Thyroid function tests should be followed carefully in preterm infants at risk for hypothyroxinemia, and treatment should be instituted promptly if indicated by elevated TSH. b. Transient hypothyroidism caused by maternal antithyroid medication will resolve spontaneously when the drug is cleared from the infant’s circulation, usually within a day or two after birth. The infant’s serum T4 and TSH should be monitored to ensure that they return to normal. Supplementation with levothyroxine is not usually necessary. c. Transplacentally acquired TRBAb can be slow to degrade completely; therefore, most infants will require supplementation for several months. d. TRBAb levels in the infant can be monitored to determine when to discontinue therapy. Breastfeeding is not contraindicated in neonates whose mothers continue their antithyroid medication in the postpartum period, as very little passes into the breast milk. G. Outcome. a. CH is one of the most preventable causes of mental retardation. Most infants with early diagnosis and early and adequate treatment will have normal IQs. A delay in treatment after birth can lower IQ by several points per week (Fisher, 2000). b. Lifelong thyroid replacement therapy is necessary for normal growth and development. c. Infants who are taking levothyroxine should not ingest soy-based products, including soy formula, because soy interferes with the absorption of levothyroxine, and the infant can remain hypothyroid (Fruzza et al., 2012). 2. Transient hypothyroidism. b. More research is needed to determine whether hypothyroxinemia of prematurity is a benign physiologic phenomenon or a cause of psychomotor and neurodevelopmental sequelae in the preterm population. 2. Rare causes of neonatal hyperthyroidism include McCune–Albright syndrome and activating mutations in the TSH receptor. B. Pathophysiology of Graves’ disease. 2. Maternal TSH receptor–stimulating antibodies (TRSAb) cross the placenta readily and stimulate the fetal thyroid gland, causing an overproduction of thyroid hormone and in some cases, development of a goiter. Usually the higher the TRSAb level in the mother, the more severely affected the infant. 3. Hyperthyroidism in the neonate is typically transient, lasting approximately 3 to 12 weeks. The clinical course varies depending on characteristics of the mother’s disease and treatment. The onset of hyperthyroidism may be delayed for a week or longer in neonates whose mothers produce not only TRSAb but TRBAb as well. 4. Similarly, if the mother took antithyroid medication during pregnancy, the neonate might not exhibit evidence of hyperthyroidism for several days until the drugs are metabolized (and might even be hypothyroid during that time). 5. Occasionally, the hyperthyroidism persists beyond the expected recovery period and becomes true, permanent Graves’ disease. C. Clinical presentation and assessment. 2. In severe cases of untreated maternal Graves’ disease, advanced bone age, craniosynostosis, and microcephaly are evident in both the fetus and newborn. 3. The infant should be examined for a goiter, which can be very small or large enough to compress the trachea and cause respiratory distress in the newborn. A goiter is a symmetrical, smooth enlargement of the gland and can be recognized as a swelling in the anterior neck of the neonate. To examine the neonate for goiter, place the infant supine and elevate the trunk while allowing the head to fall back gently (LynShue and Witchel, 2007). D. Diagnostic studies. 2. A TRSAb titer in the neonate will give an indication of the expected severity of the disease course. Infants at risk (e.g., high maternal titer of TRSAb) for severe thyrotoxicosis require frequent monitoring of free T4 and TSH. 3. A thorough maternal history is essential (e.g., history of radioablation therapy, antithyroid drugs taken during pregnancy and when they were taken, and maternal symptoms, if any). E. Management. 2. Propranolol can be used to manage cardiovascular symptoms. The infant’s serum T4 must be followed closely during treatment for possible iatrogenic hypothyroidism. TRSAb levels are also followed to monitor the infant’s recovery and aid in determining the appropriate time for weaning antithyroid medication. 3. In cases of maternal thyrotoxicosis, thyrotoxicosis should be anticipated in the neonate so diagnosis and treatment can be instituted without delay. F. Complications. G. Outcome. 1. Neonatal hyperthyroidism is almost always transient. 2. The mortality rate of 12% to 16% is related to cardiovascular compromise, arrhythmia, tachycardia, and heart failure (Peters and Hindmarsh, 2007). 3. Survivors of severe, prolonged thyrotoxicosis often have permanent neurologic impairment from premature craniosynostosis and the direct effects of excess thyroid hormones on the brain. 2. The adrenal medulla produces and stores catecholamines (epinephrine, norepinephrine, dopamine) and is linked to the sympathetic nervous system. 3. All adrenocortical hormones (steroids) are synthesized from cholesterol. Three classes of steroids are produced by the adrenal cortex: glucocorticoids, mineralocorticoids, and androgens. B. Adrenocortical hormones. 2. Cortisol is closely regulated by ACTH and the hypothalamic–pituitary–adrenal axis via an acute or chronic negative-feedback loop. Increased plasma cortisol inhibits secretion of corticotropin-releasing hormone and ACTH, whereas decreased plasma cortisol permits their release. Cortisol is also released in response to stress, hypoglycemia, surgery, extreme heat or cold, decreased oxygen concentration, infection, or injury. 3. Aldosterone, the most important mineralocorticoid, regulates renal sodium (Na+) and water retention and potassium (K+) excretion, thus affecting not only electrolyte balance but also blood pressure and intravascular volume. Aldosterone is regulated by the plasma renin–angiotensin system and by plasma K+ concentrations. A drop in intravascular volume or the Na+ concentration or a rise in the K+ level stimulates the renin–angiotensin system, which in turn stimulates production of aldosterone. 4. Adrenal androgens include dehydroepiandrosterone (DHEA), DHEA sulfate, and androstenedione and are regulated by ACTH. These steroids have minimal androgenic activity but are converted in the peripheral tissues to two more potent androgens: testosterone and dihydrotestosterone. C. Fetal adrenocortical development. 2. The fetal adrenal gland and the placenta are an integrated endocrine organ known as the fetoplacental unit. The fetal zone, deficient in a critical enzyme for cortisol synthesis, produces mostly DHEA and DHEA sulfate. These are the precursors for placental estrogen, which is vital to maintenance of the pregnancy and fetal well-being. In turn, the placenta provides substrates for fetal cortisol production. 3. Until about 30 weeks of gestation, fetal cortisol comes from both the fetal gland and transplacental transfer. In the placenta, 80% of maternal cortisol is rapidly metabolized to inactive cortisone to protect the fetus from very high cortisol levels. Near term, maturation of fetal enzyme systems allows greater conversion of cortisone back to cortisol and synthesis of cortisol from cholesterol. Increases in circulating cortisol during the last 10 weeks of gestation (the prenatal cortisol surge) induce critical physiologic changes that prepare the fetus for extrauterine life, including maturation of pulmonary surfactant. 4. Aldosterone production increases throughout pregnancy, preparing the fetus to assume control of salt and water balance after birth. D. Neonatal adrenocortical function. 2. A diurnal pattern of cortisol secretion is evident at about 2 to 3 months of age in most infants, influenced more by environmental factors than genetic factors (Custodio et al., 2007). Cortisol in the newborn plays a key role in response to stress and illness, and is important in the maintenance of blood pressure (Ng, 2011). 3. Aldosterone concentration and plasma renin activity (PRA) are elevated compared with values for older infants, allowing for positive Na+ balance until the kidneys fully mature. The hyponatremia and urinary Na+ loss often seen in preterm infants during the early postnatal weeks are due to a relative mineralocorticoid deficiency as a consequence of immaturity of both the kidneys and the adrenal glands. A. Transient adrenocortical insufficiency of prematurity. 2. Manifestations are a low serum cortisol, normal or exaggerated pituitary response, and good recovery of adrenal function by day 14 of life (Ng, 2011). 3. Some very low birth weight infants with inotrope and volume-resistant hypotension show an inadequate adrenal response to stress in the immediate postnatal period (Ng, 2011). 4. Whether a relative adrenal insufficiency contributes to hemodynamic instability and hypotension in critically ill infants is still under debate (Nimkarn and New, 2012). B. Adrenal hemorrhage. 2. Although often asymptomatic, classic findings include jaundice, pallor, and a flank mass on either side (although hemorrhage is more common on the right) with discoloration and purpura of the overlying skin and discoloration of the scrotum (Mutlu et al., 2011). In severe cases, the infant may exhibit signs of adrenal insufficiency. Small hemorrhages can be initially undetected, but eventually manifest in anemia and persistent jaundice (Janjua and Batisky, 2012). 3. Adrenal hemorrhage can be visualized on ultrasound, and usually resolves in 4 to 16 weeks (Mutlu et al., 2011). A. Definition: A group of autosomal recessive genetic disorders resulting from deficient activity of one of the enzymes required to synthesize cortisol from cholesterol in the adrenal cortex. Each enzyme is encoded by its own gene. Mutation of the 21-hydroxylase (21-OHD) gene, CYP21, accounts for 95% of disorders, and is the most common cause of ambiguous genitalia in the neonate. Currently there are 127 known different mutations of CYP2A12 that range from having absolutely no enzyme activity to partial enzyme function. The worldwide incidence of classic 21-OHD is approximately 1 in 5000 to 1 in 15,000 live births (Witchel and Azziz, 2011; Stokowski, 2014). B. Pathophysiology (Fig. 30-2): 2. Lack of fetal 21-OHD prevents conversion of progesterone to its two end products: cortisol and aldosterone. 3. By reduced negative-feedback regulation, the absence of cortisol causes oversecretion of ACTH, which chronically stimulates the adrenal cortex, resulting in hyperplasia. 4. The cortisol precursor 17-OHP accumulates in the blood because its conversion to cortisol is blocked. 5. The excess 17-OHP enters the unblocked androgen metabolic pathway, which results in an overproduction of androgens. At a critical stage in fetal development, androgens cause virilization of the external genitalia in female fetuses. Also important may be the effects of this androgen exposure on the developing CNS.
Endocrine Disorders
THE ENDOCRINE SYSTEM
Endocrine Gland
Hormones Produced
Hypothalamus
Corticotropin-releasing hormone (CRH)
Thyrotropin-releasing hormone (TRH)
Gonadotropin-releasing hormone (GnRH)
Somatostatin
Growth hormone–releasing hormone (GHRH)
Prolactin-releasing factor (PRF)
Prolactin release–inhibiting hormone (PIH; dopamine)
Anterior pituitary
Adrenocorticotropic hormone (ACTH)
Thyroid-stimulating hormone (TSH; thyrotropin)
Follicle-stimulating hormone (FSH)
Growth hormone (GH)
Luteinizing hormone (LH)
Prolactin (PRL)
Posterior pituitary
Antidiuretic hormone (ADH; arginine vasopressin)
Oxytocin (OCT)
Thyroid gland
Thyroxine (T4)
Triiodothyronine (T3)
Calcitonin
Parathyroid gland
Parathyroid hormone (PTH)
Adrenal medulla
Epinephrine (adrenaline)
Norepinephrine (noradrenaline)
Adrenal cortex
Cortisol (hydrocortisone)
Aldosterone
Pancreas
Insulin
Glucagon
Somatostatin
Pineal gland
Melatonin
PITUITARY GLAND DISORDERS
THYROID GLAND DISORDERS
The Thyroid Gland
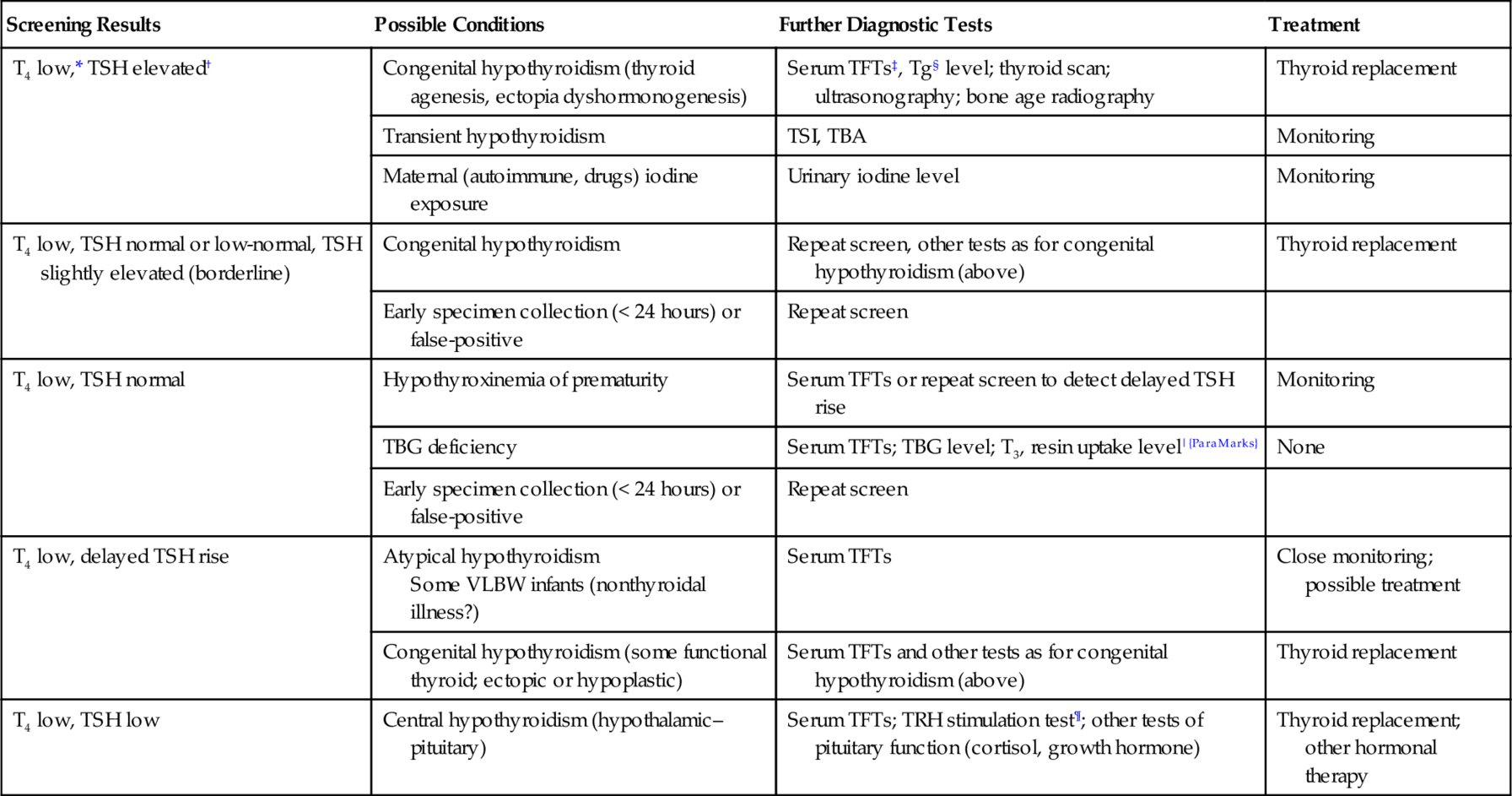
Hypothyroidism
Screening Results
Possible Conditions
Further Diagnostic Tests
Treatment
T4 low,* TSH elevated†
Congenital hypothyroidism (thyroid agenesis, ectopia dyshormonogenesis)
Serum TFTs‡, Tg§ level; thyroid scan; ultrasonography; bone age radiography
Thyroid replacement
Transient hypothyroidism
TSI, TBA
Monitoring
Maternal (autoimmune, drugs) iodine exposure
Urinary iodine level
Monitoring
T4 low, TSH normal or low-normal, TSH slightly elevated (borderline)
Congenital hypothyroidism
Repeat screen, other tests as for congenital hypothyroidism (above)
Thyroid replacement
Early specimen collection (< 24 hours) or false-positive
Repeat screen
T4 low, TSH normal
Hypothyroxinemia of prematurity
Serum TFTs or repeat screen to detect delayed TSH rise
Monitoring
TBG deficiency
Serum TFTs; TBG level; T3, resin uptake level|{ParaMarks}
None
Early specimen collection (< 24 hours) or false-positive
Repeat screen
T4 low, delayed TSH rise
Atypical hypothyroidism
Some VLBW infants (nonthyroidal illness?)
Serum TFTs
Close monitoring; possible treatment
Congenital hypothyroidism (some functional thyroid; ectopic or hypoplastic)
Serum TFTs and other tests as for congenital hypothyroidism (above)
Thyroid replacement
T4 low, TSH low
Central hypothyroidism (hypothalamic–pituitary)
Serum TFTs; TRH stimulation test¶; other tests of pituitary function (cortisol, growth hormone)
Thyroid replacement; other hormonal therapy
Hyperthyroidism
ADRENAL GLAND DISORDERS
The Adrenal Gland
Adrenal Disorders in the Neonate
Congenital Adrenal Hyperplasia
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


30: Endocrine Disorders
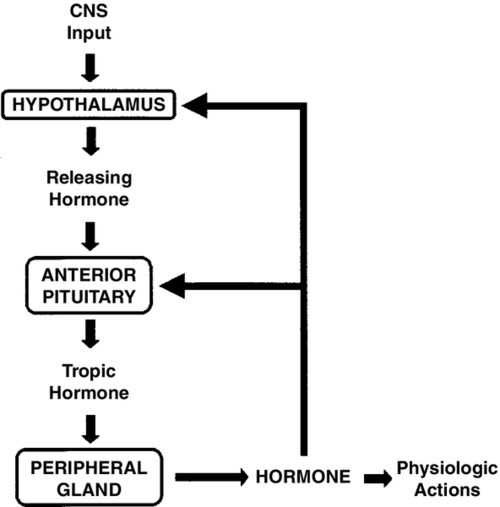
FIGURE 30-1 ■ Negative-feedback-loop control of endocrine gland function. Hypothalamic releasing hormones stimulate pituitary trophic hormones, which in turn act on peripheral glands to release hormones. Levels of circulating hormones then exert feedback control on the pituitary and hypothalamus, modulating further output by these glands. CNS, Central nervous system.
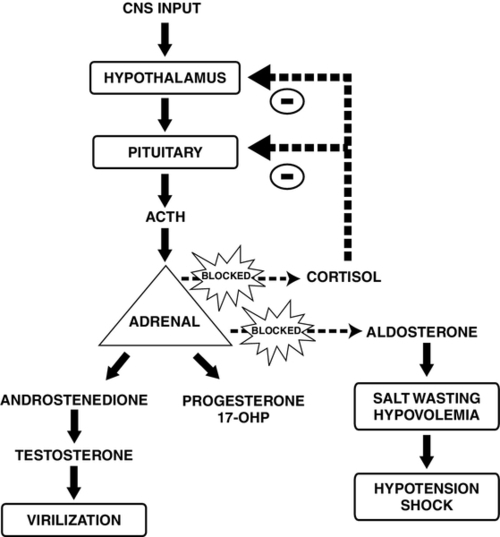
FIGURE 30-2 ■ Pathophysiology of congenital adrenal hyperplasia caused by 21-hydroxylase deficiency. In the absence of cortisol, adrenocorticotropic hormone (ACTH) stimulates the adrenal cortex to produce virilizing androgens. Diminished production of aldosterone leads to salt wasting and hypovolemia. CNS, Central nervous system; 17-OHP, 17-hydroxyprogesterone. (Adapted from Martin, R.J., Fanaroff, A.A., and Walsh, M.C.: Fanaroff and Martin’s neonatal-perinatal medicine: Diseases of the fetus and infant [9th ed.]. Philadelphia, 2011, Elsevier Mosby.)
