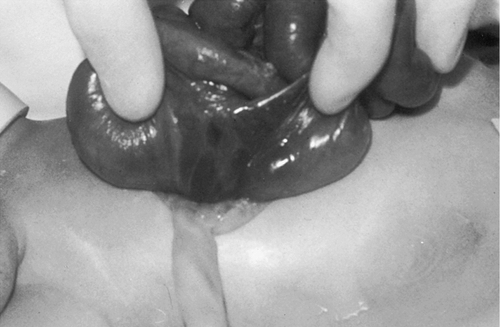
CHAPTER 29 1. Discuss normal and abnormal abdominal assessment findings. 2. Discuss common laboratory and diagnostic tests used to evaluate the gastrointestinal system. 3. Differentiate between omphalocele and gastroschisis. 4. Identify four common associations in infants with intestinal obstruction. 5. Identify the clinical presentation of a neonate with tracheoesophageal fistula. 6. Describe radiographic findings in an infant with duodenal atresia. 7. Identify one gastrointestinal disorder that is considered a surgical emergency. 8. Identify the gastrointestinal presentation of infants with cystic fibrosis. 9. Describe the defect in Hirschsprung’s disease. 10. Identify the three mechanisms involved in the pathogenesis of necrotizing enterocolitis. 11. Describe the clinical presentation of an infant with biliary atresia. 12. Identify at least three management strategies for the infant with cholestasis. 13. Identify at least three management strategies for the infant with gastroesophageal reflux. 14. Identify the triad of anomalies occurring in prune-belly syndrome. 15. Describe the symptoms of diaphragmatic hernia. 16. Differentiate between unconjugated and conjugated bilirubin. 17. Compare and contrast physiologic and nonphysiologic jaundice. 18. Describe management of an infant receiving phototherapy. 19. Define “hydrops.” Unique embryologic features of the gastrointestinal (GI) tract, such as the obliteration and recanalization of the GI tract, midgut herniation into the umbilical cord, and rotation of the intestines, make the GI tract prone to a variety of congenital anomalies. Anomalies may affect any part of the GI tract, from the mouth to the anus. Atresias, stenoses, and functional obstructions account for the vast majority of congenital defects. As for acquired defects, necrotizing enterocolitis (NEC) is the most common serious GI illness in neonates. This chapter will review the more common GI anomalies and a variety of multisystem disorders that have significant GI involvement, such as prune-belly syndrome, congenital diaphragmatic hernia (CDH), hyperbilirubinemia, and hydrops. B. Week 4. Intestine is present; esophagus and trachea separate and are distinct; stomach becomes obvious; liver is present. C. Week 5. Esophagus, stomach, proximal duodenum present; intestine elongates into a loop and begins to rotate. D. Week 6. Stomach rotates into adult position; distal duodenum, jejunum, ileum, cecum, ascending colon, and proximal two thirds of transverse colon present; rapidly developing midgut herniates into umbilical cord. E. Week 7. Rapid endothelial proliferation results in temporary duodenal occlusion; intestinal loops herniate into umbilical cord, lengthen, and rotate; and urorectal septum fuses with cloacal membrane, separating rectum from the developing urinary bladder. F. Week 8. Small intestine recanalizes; intestinal villi develop; diaphragm complete. G. Weeks 9 and 10. Intestines begin to reenter abdominal cavity and continue counterclockwise rotation around the axis of the superior mesenteric artery. H. Week 12. Muscular layers of intestine are present; active transport of amino acids begins; pancreatic islet cells appear; bile appears; lactase appears. I. Week 16. Meconium is present; swallowing is observed. J. Week 24. Ganglion cells are detected in the rectum. K. Week 26. Random peristalsis begins. L. Weeks 34 to 36. Sucking and swallowing become coordinated. M. Weeks 36 to 38. Maturity of GI system completed. N. Weeks 5 to 40. Intestine elongates approximately 100-fold (small intestine is 6 times the length of the colon). A. Absorption and digestion of nutrients. B. Elimination of waste products. C. Maintenance of fluid and electrolyte balance. D. Protection of host from toxins and pathogens. 1. Family: presence of GI disease. 2. Presence of genetic syndrome: Major syndromes associated with GI defects include the following (Jones et al., 2013): a. Apert’s syndrome: pyloric stenosis, tracheoesophageal fistula/esophageal atresia (TEF/EA), CDH. b. Beckwith–Wiedemann syndrome: umbilical defects, diastasis recti, posterior diaphragmatic eventration, pancreatic hyperplasia resulting in hypoglycemia. c. Fetal hydantoin syndrome: umbilical hernia, duodenal atresia. d. Meckel–Gruber syndrome: liver defects (bile duct proliferation, fibrosis, cysts), single umbilical artery, patent urachus, omphalocele, intestinal malrotation, imperforate anus. e. Sirenomelia: imperforate anus, anal agenesis. f. Trisomy 13: umbilical defects, intestinal malrotation, CDH. g. Trisomy 18: umbilical defects, pyloric stenosis, intestinal malrotation, TEF/EA, CDH, single umbilical artery. h. Trisomy 21: Hirschsprung’s disease, pyloric stenosis, duodenal atresia, intestinal malrotation. i. VATERR association (vertebral defects, imperforate anus, tracheoesophageal fistula and/or esophageal atresia, and radial and renal dysplasia). Additional: single umbilical artery. j. VACTERL association (vertebral defects, anal atresia, cardiac abnormalities, tracheoesophageal fistula and/or esophageal atresia, renal agenesis or dysplasia, and limb defects). 3. History of present illness. (1) Abdomen can be seen by 10 weeks of gestation, stomach by 13 weeks. (2) Abdomen can be assessed for intactness of abdominal wall, umbilical cord insertion, stomach as fluid-filled chamber, bowel dilation, or indication of obstruction. (3) Polyhydramnios (> 2000 mL) may indicate interference with fetal swallowing or intestinal obstruction. (4) Oligohydramnios (< 500 mL) may indicate renal agenesis or dysgenesis. b. Birth weight, weight loss/gain, reflux, gastric aspirates, vomiting (bilious vs. nonbilious, projectile), timing of passage of first meconium stool, stool color, presence of blood, diarrhea, constipation, abdominal distention or tenderness, jaundice (Seidel et al., 2010). B. Abdominal assessment (Goodwin, 2009; Seidel et al., 2010). (1) Normal: rounded, soft, symmetrical, pink. (2) Abnormal: erythematous, dusky, mottled. (3) Distended: intestinal obstruction, infection, organomegaly, ascites. (4) Scaphoid: associated with CDH. (5) Asymmetrical: mass, organomegaly, intestinal obstruction. b. Muscular development. (1) Flat, flabby, lumpy: prune-belly syndrome. (2) Gap between rectus muscles: diastasis recti. (3) Externalization of abdominal contents: omphalocele, gastroschisis, bladder exstrophy. (4) Hernias: protrusions of an organ or tissue through an abnormal opening. Common in three areas: (b) Inguinal. (i) More common in males, especially very low birth weight. (ii) Frequently bilateral. (iii) May not be evident until second or third month of life. (iv) Usually readily reducible. (c) Femoral. (i) Rare but more common in females. (ii) Located just below inguinal ligament on anterior aspect of thigh. c. Umbilicus/umbilical cord. (2) Normally three vessels: two ventrally situated arteries, one dorsally situated vein. (3) Usually dries and spontaneously detaches in 10 to 14 days. (4) Green or yellow staining suggests in-utero meconium passage. (5) Wet, foul-smelling cord, or periumbilical redness: omphalitis. (6) Persistent clear drainage: patent urachus. (7) Ileal fluid drainage: omphalomesenteric duct. (8) Serous or serosanguineous drainage: granuloma. (9) Abnormally thick: single herniated loop of intestine. (10) Thick, gelatinous: large for gestational age. (11) Thin, small: intrauterine growth restriction. d. Bowel loops. (2) Presence: obstruction. e. Movements. (1) Should move in synchrony with respirations. (2) Movements not in synchrony may represent respiratory distress, peritoneal irritation, or central nervous system (CNS) disease. (3) Peristalsis: not normally seen. (a) May be seen in premature infants with thin abdominal walls. (b) Presence: associated with hypertrophic pyloric stenosis. f. Veins. (1) Superficial veins become more prominent with abdominal distention. (2) Dilated veins: venous obstruction. g. Perineum: inspected for patency of anus and presence of fistulas. 2. Auscultation. a. Done before palpation (to avoid altering sounds). b. Bowel sounds. (1) Become audible within 15 to 30 minutes after birth (Montrawl, 2014). (2) Should have a metallic clicking quality. (3) Hyperactive or hypoactive does not necessarily represent pathologic change. Other historical or clinical findings should be taken into consideration when interpreting bowel sounds. (4) Increased sounds. (b) Hirschsprung’s disease. (c) Diarrhea. (5) Decreased or absent sounds. (b) Starvation. (c) Medication administration: opiates, magnesium, anesthesia. c. Vascular sounds: bruit, similar to murmur. Caused by turbulent blood flow through an artery, especially if heard despite position change of infant. Bruits may indicate partial vascular obstruction. d. Friction rub: peritoneal inflammation, splenic involvement, hepatic tumor, abscess. 3. Percussion. b. Two main sounds to listen for: (1) Tympanic: low-pitched, heard over gas-filled structures (stomach). (2) Dullness: high-pitched, short, heard over dense or solid organs (liver, spleen). 4. Palpation. (2) Organ position. (a) Liver should be 1 to 2 cm below right costal margin, midclavicular line. (b) Spleen rarely palpable. Tip should not be more than 1 cm below left costal margin. (c) Kidneys are about 4 to 5 cm in length. Right kidney is easier to palpate and is lower than left. (3) Organomegaly. (4) Masses. (5) Pulsations. (6) Fluid. b. Technique: start in lower quadrants and progress to upper quadrants using a bimanual approach. Place one hand under infant’s back; using free hand, start with light palpation, then progress to deep palpation. c. Hints to relax infant. (2) Flex legs. (3) Use gentle circular motion. (4) Slowly increase depth of palpation. (5) Palpate any known areas of tenderness last. C. Diagnostic tests. a. Gastric aspirate. Measure pH of gastric contents. b. Apt test. (2) Can be done on gastric fluid or stool. 2. Stool examination. a. Normal: initially meconium followed by soft, yellow stool after milk introduction. b. Examined for color, consistency, odor, blood, mucus, pus, tissue fragments, bacteria, and parasites. c. Color may be influenced by diet, dyes, drugs, pathology. (1) Green: indomethacin, meconium. (2) Greenish black: iron, meconium. (3) Black: iron, swallowed blood, blood from high GI tract lesion. (4) Pale: biliary atresia. (5) White: antacids, barium. (6) Red: bright red indicates blood from low GI tract lesion or anal fissure; currant jelly indicates intussusception. d. Odor. (2) Purulent odor suggests colitis. e. CHO malabsorption. (1) pH less than 5 in infants is suggestive. (2) Reducing substance test (Clinitest Reagent Tablets, Bayer Corp., Diagnostic Division, Elkhart, Ind.) detects CHO malabsorption indicating impaired bowel mucosal integrity or dysfunction that may be seen with NEC (Pinheiro et al., 2003). f. Guaiac. 3. pH probe test: 24-hour pH probe study to diagnose GI reflux. Detects acid reflux only; does not detect nonacid (milk) or gas reflux. 4. Radiologic studies. a. Plain radiograph (see Chapter 14). (1) Bowel gas pattern (Montrawl, 2014). (a) At birth, GI tract is fluid-filled and gasless. (b) Within 30 minutes, gas should be in stomach. (c) By 3 to 4 hours, gas should be in small intestine. (d) After 6 to 8 hours, gas should be in entire intestine. (2) Absence of gas below pylorus: possible indication of obstruction. b. Upper GI series: done to assess structure and function of esophagus and evaluate for pyloric stenosis, malrotation, and strictures. (2) Water-soluble products are preferred in cases of suspected perforation. c. Lower GI series: may be used to detect presence of malrotation, Hirschsprung’s disease, meconium ileus, and meconium plug syndrome. May be therapeutic in meconium ileus and meconium plug syndrome. 5. Ultrasonography: may be used to diagnose suspected cases of pyloric stenosis, duplications, gastroesophageal reflux (GER), or biliary atresia. 6. Scintigraphy (nuclear scan): used to evaluate gastric emptying, aspiration with swallowing, reflux with aspiration, and liver excretory function. Radionuclide-tagged formula is fed to the infant and recorded by a gamma camera. 7. Endoscopy: used to directly visualize the upper or lower GI mucosa. Endoscopic retrograde cholangiopancreatography (ERCP) visualizes the biliary pancreatic ducts. 8. Fecal fat: used to diagnosis malabsorption syndromes. Fecal fat content greater than 6 g/24 hours is associated with malabsorption syndrome. 9. Culture: helpful in differentiating bloody diarrhea caused by infection versus hypoxic insult to intestine. D. Laboratory tests (Table 29-1) (Kee, 2005). a. Synthesized in the liver; it is the most abundant plasma protein. b. Decreased levels occur in hepatocellular injury; usually a late finding. 2. Alkaline phosphatase (ALP). b. Elevated levels occur in obstructive liver disease (e.g., biliary atresia) and hepatitis, as well as in bone disease. 3. Aminotransferase activity. b. Aspartate aminotransferase (AST) catabolizes the reversible transfer of the α-amino group of aspartic acid to the α-keto group of α-ketoglutaric acid, leading to the formation of oxaloacetic acid. c. ALT and AST are the most sensitive tests of hepatocyte necrosis. ALT is more specific than AST because it is not found in high concentrations in other tissues. d. High elevations occur in hepatocellular injury. Slight elevations occur in cholestasis. Serum ALT greater than 300 units coupled with jaundice signals a liver disorder and not a hemolytic disorder. e. ALT-to-AST ratio is often performed to help differentiate types of liver disease. 4. Ammonia. b. Elevated in liver failure. c. Elevations occur in acute or chronic liver disease. 5. Bilirubin. a. By-product of heme breakdown. b. Increased indirect bilirubin occurs when liver function is reduced (prematurity, injury) or when there is an excessive load of unconjugated bilirubin (hemolysis). c. Increased direct bilirubin occurs when the liver cannot excrete conjugated bilirubin into the bile ducts or biliary tract (biliary atresia, cholestasis). 6. Prothrombin time (PT). a. Measures the time required for prothrombin (factor II) to be converted to thrombin. b. In cases of obstructive liver disease in which bile acids do not reach the intestine, fat-soluble vitamins are not absorbed (required for coagulation factors). c. Prolonged PT occurs in patients with hepatocellular injury and biliary obstruction. 7. International Normalized Ratio (INR): ratio of a patient sample PT to a normal PT. 8. Serum bile acids. b. Elevations occur in acute and chronic liver disease. 1. Definition: central defect with herniation of the abdominal viscera into the umbilical cord covered by a thin, avascular membranous sac composed of amnion and peritoneum with a small amount of Wharton’s jelly. Umbilical arteries and veins insert into the apex of the defect (Fig. 29-1). 2. Etiology: multifactorial, including environmental, chromosomal (recessive and dominant forms), and folding abnormality of the germ disc, with failure to close the ventral abdominal wall. 3. Incidence: 2.5 per 10,000 live births. 4. Associated conditions: many will have associated anomalies or be part of a syndrome. a. Prematurity; small for gestational age. b. Cardiac defects (50%). c. Intestinal malrotation and/or atresia. d. Pentalogy of Cantrell: upper abdominal omphalocele above the umbilical cord, diaphragmatic hernia, sternal cleft, pericardial defect, ectopia cardiac defect. e. Neurologic anomalies, neural tube (40%). f. Genitourinary anomalies. g. Skeletal anomalies. h. Chromosomal anomalies (50% to 65%). Common anomalies include trisomy 13, 18, and 21, with 18 being the most frequent. Frequent in infants with Beckwith–Wiedemann syndrome (chromosome 11). 5. Diagnosis. a. Maternal serum markers: elevated serum α-fetoprotein to detect open defects. b. High-resolution prenatal ultrasound. c. Amniocentesis: chromosome analysis for associated defects; amniotic fluid α-fetoprotein and acetylcholinesterase levels to evaluate for defects associated with abnormal values. d. Inspection at birth. (2) Large defect: may contain intestines, stomach, liver, spleen, bladder, uterus and ovaries, testicles; deficient upper musculature of umbilical ring. (3) The thin, avascular membranous sac may rupture before or at time of delivery; must be differentiated from gastroschisis because of the high rate of associated anomalies with omphalocele. 6. Prognosis. a. Mortality rate is related to size of defect and severity of associated anomalies. B. Gastroschisis (Sadler and Rasmussen, 2010). 1. Definition: herniation of abdominal contents through an abdominal wall defect lateral to the umbilical ring; umbilical ring and cord are normal; right-sided predominance (Fig. 29-2). See Table 29-2 for a comparison of omphalocele and gastroschisis. TABLE 29-2 Comparison of Omphalocele and Gastroschisis IUGR, Intrauterine growth restriction.
Gastrointestinal Disorders
GASTROINTESTINAL EMBRYONIC DEVELOPMENT
FUNCTIONS OF THE GASTROINTESTINAL TRACT
ASSESSMENT OF THE GASTROINTESTINAL SYSTEM
ABDOMINAL WALL DEFECTS (CHRISTISON-LAGAY ET AL., 2011; ISLAM, 2012; LEDBETTER, 2012)
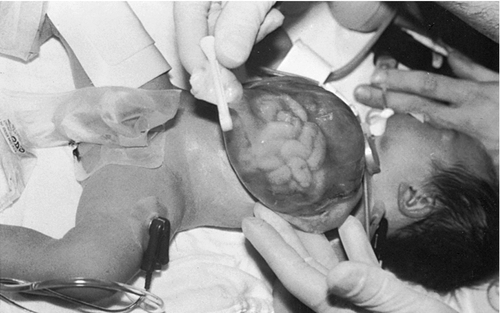
Omphalocele
Gastroschisis
Incidence
1:5000 to 1:6000
1:30,000 to 1:50,000
Covering
Present, may be ruptured
None
Site
Umbilical
Paraumbilical, usually to the right
Fascial defect
Small or large
Small
Herniated organs
Intestines; stomach, liver, spleen sometimes
Intestines; rarely liver
Appearance of herniated bowel
Normal, unless sac is ruptured
Often edematous, matted
Associated anomalies
45% to 55%
10% to 15%
IUGR
Less common
Common
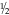
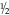
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


