CHAPTER 28 2. Discuss implementation of a universal pulse oximetry screening protocol. 3. Name the major classifications of congenital heart disease; list two anomalies in each. 4. Discuss the link between congenital heart disease and genetics, including use of available tests. 5. Define and describe the anatomy, clinical manifestations, and the possible medical and/or surgical treatment of tetralogy of Fallot (TOF), coarctation of the aorta, patent ductus arteriosus (PDA), ventricular septal defect (VSD), atrial septal defect (ASD), and hypoplastic left heart syndrome (HLHS). 6. Describe the basic medical rationale for care of the newborn infant with a suspected or identified cardiac defect. 7. Describe the basic surgical rationale for treatment of major cardiovascular defects. 8. List signs and symptoms of congenital heart abnormalities in the newborn infant. 9. Discuss available diagnostic modalities and the congenital cardiac defects they are used to diagnose. 10. List the signs and symptoms of PDA and the current medical, transcatheter, and surgical interventions. 11. Discuss the treatment modalities used in congestive heart failure, including the risks and benefits to the neonate. 12. Define the three classifications of shock, and list one cause under each category. 13. Discuss the major complications and sequelae of open heart surgery and list two factors that contribute to the sequelae. Until the 20th century, there was limited clinical interest in congenital heart disease. In the majority of cases, the cardiac anomaly was incompatible with life; in the others, no treatment existed either to remedy the condition or to relieve its symptoms. Today, with advances in prenatal and early postnatal diagnosis and treatment of congenital heart defects (CHDs), the survival and quality of life of infants with congenital heart disease have markedly improved. Yet, anywhere from 20% to 30% of neonates with critical heart defects are not diagnosed prior to discharge from the hospital following birth (Mahle et al., 2009). A delay in diagnosis leads to increased morbidity and mortality (Altman, 2013). Due to the prevalence of missed diagnosis before discharge, the U.S. Department of Health and Human Services made a recommendation for universal pulse oximetry screening prior to discharge from the hospital. The American Academy of Pediatrics Section on Cardiology and Cardiac Surgery endorsed the recommendation in 2012. Advances in fetal echocardiography have resulted in a significant increase in the prenatal diagnosis of many CHDs, allowing for extensive parental counseling, thus providing time for parents to make decisions about treatment options prior to birth (Dorfman et al., 2008; Kaplan et al., 2005) and delivery at a tertiary hospital (Donofrio et al., 2013). Telemedicine has also made a big impact on CHD, allowing for accurate diagnosis at remote hospitals, thus quicker implementation of care (Grant et al., 2010). In addition, advancements in angiography, interventional catheterization, and the advent of three-dimensional (3-D) echocardiography (Friedberg et al., 2010), coupled with a more complete understanding of newborn physiology, have led to improvements in diagnostic and treatment capabilities with decreased risk. In addition, newer surgical techniques that limit time spent in deep hypothermia with circulatory arrest or on continuous cardiopulmonary bypass, use of normothermic cardiopulmonary bypass, and surgery on beating hearts not only permits the total correction of many forms of CHDs in the neonatal period but increases survival and decreases morbidities (Newazhay et al., 2012). In most cases, early intervention is essential to minimize and prevent long-term morbidity or early death (Knowles et al., 2012). Whereas these advances have allowed for correction of many CHDs with dramatic decreases in mortality, neurodevelopmental morbidities are still found (Stone, 2010). Multiple clinical trials reported in the literature substantiate earlier findings of neurodevelopmental impairments in patients following neonatal cardiac surgery (Newburger et al., 2012). Although overall intelligence has been spared, only 21% of school-age patients function at age-appropriate levels, 37% have moderate disabilities, and 6% have severe disabilities. The disabilities were identified as motor and cognitive impairments, including speech and language delays, gross and fine motor deficiencies, and visual–motor delays. Many studies have linked these outcomes to deep hypothermic cardiac arrest and continuous cardiopulmonary bypass that is used during open heart surgery. Fetal cardiac development occurs rapidly from day 18 to the 12th week of fetal life (Sedmera, 2011). 1. Heart development is first identified at 18 to 19 days of fetal life with formation of a heart tube (Table 28-1; Fig. 28-1, A). TABLE 28-1 Timeline for Fetal Heart Development Data from Collins-Nakai, R. and McLaughlin, P.: How congenital heart disease originates in life. Cardiology Clinics, 20(3):367-383, 2002; Park, M.K.: Pediatric cardiology for practitioners (5th ed.). Philadelphia, 2008, Elsevier Mosby; Theorell, C.: Cardiovascular assessment of the newborn. Newborn and Infant Nursing Reviews, 2(2):111-127, 2002. 2. The heart tube elongates and develops dilatations and contractions that will later form the ventricles, bulbus cordis, and outflow tracts (Fig. 28-1, B). Molecular studies are beginning to identify that the heart tube actually becomes the left ventricle, but more studies are needed (van de Berg and Moorman, 2009). 3. The pairs of aortic arches develop from the cephalic, extracardial portion of the heart tube. The caudal end will form the early ventricle (Fig. 28-1, C). 4. Abnormal development during this time will include corrected transposition and dextrocardia (Collins-Nakai and McLaughlin, 2002). B. Cardiac septation: begins in the middle of the fourth week and is complete by the end of the fifth week of fetal life (Fig. 28-1, D). 1. Atrial septum and foramen ovale are formed from two septa and endocardial cushions. b. A second septum (septum secundum) appears to the right of the septum primum and grows to overlap the ostium secundum. Thus the flapped opening, the foramen ovale, is created (Fig. 28-2). 2. Ventricular septation results from fusion of the endocardial cushions and dilation and fusion of the ventricles (Fig. 28-3). 3. Tissue of endocardial cushion forms the atrioventricular valves (Fig. 28-4). a. Tricuspid valve: between right atrium and ventricle. b. Mitral valve: between left atrium and ventricle. 4. Abnormal development during cardiac septation can lead to the following: a. VSD, ASD, and endocardial cushion defect (atrioventricular canal). b. Absence, deformation, stenosis, or atresia of the tricuspid and/or mitral valves. A. Single vessel (truncus arteriosus) extends from the ventricles until the fourth week of fetal life. It will then separate into the aorta and the pulmonary artery (Fig. 28-5). B. Development of aorta and aortic branches (Table 28-2). The truncus arteriosus connects with the aortic sac, which in turn connects with the paired aortic branches. A series of six paired aortic arches then form in succession (Fig. 28-6). 1. The third arches become part of the common carotid arteries. 2. The right fourth arch becomes the proximal portion of the right subclavian artery. 3. The left fourth arch becomes the aortic arch segment between the left common carotid artery and left subclavian artery. 4. The left sixth arch becomes part of the left pulmonary artery and the ductus arteriosus. 5. The right sixth arch forms the right pulmonary artery. 6. Abnormalities include interrupted aortic arch and coarctation of the aorta. TABLE 28-2 Development of Aorta and Aortic Branches Data from Collins-Nakai, R. and McLaughlin, P.: How congenital heart disease originates in life. Cardiology Clinics, 20(3):367-383, 2002; Park, M.K.: Pediatric cardiology for practitioners (5th ed.). Philadelphia, 2008, Elsevier Mosby; Theorell, C.: Cardiovascular assessment of the newborn. Newborn and Infant Nursing Reviews, 2(2):111-127, 2002. C. Developmental abnormalities of truncal septation include (Collins-Nakai and McLaughlin, 2002): 2. TOF. 3. Pulmonary and/or aortic valve atresia or stenosis. 4. Transposition of the great vessels (dextroposition). 5. Double-outlet right ventricle. B. Electrical conduction system is functional around 10 weeks, with normal sinus rhythm seen by 16 weeks (Blackburn, 2013). C. Establishment of fetal circulation: With completion of atrial and ventricular septation, development of valves between chambers and outflow tracks, and separation of the truncus arteriosus into the great vessels, fetal circulation is established. D. Fetal circulation is anatomically and physiologically different from adult circulation, and adaptation after birth is necessary (Blackburn, 2013). For a full discussion of fetal circulation and cardiopulmonary adaptation at birth, see Chapter 4. A. Normal circulation (see Fig. 4-2). 2. As the blood flows through the lungs, it gives up carbon dioxide and gains oxygen. 3. Oxygen-rich blood returns from the lungs through the pulmonary veins. It enters the left atrium and then passes through the mitral valve into the left ventricle, which pumps it through the aortic valve and into the aorta. 4. The aorta then delivers oxygenated blood to all body organs and tissues. B. Cardiac depolarization 2. Shortening of muscle fibers (contraction) usually follows cardiac depolarization. Strength of cardiac (ventricular) contraction is measured by blood pressure or arterial pulse palpation. 3. Cardiac electrical activity does not ensure adequate cardiac function. b. Electrolyte disturbance (i.e., altered fluid composition surrounding the cells) can affect electrical activity. (1) Hypokalemia and hyperkalemia. (2) Hypocalcemia and hypercalcemia. (3) Hypoxia. (4) Acidosis. C. Cardiac output. 2. Cardiac output = stroke volume × heart rate (in liters per minute). a. As heart rate fluctuates, so will cardiac output. (1) Normal term newborn heart rate is 110 to 166 beats per minute (bpm), although it may range from 70 to 180 bpm on an individual basis (Dawson et al., 2010). b. Tachycardia seems to be an effective mechanism for improving cardiac output as long as the tachycardia does not compromise diastolic filling time and decrease coronary artery perfusion. 3. Stroke volume is relatively fixed at 1.5 mL/kg and is affected by three factors: preload, contractility, and afterload. a. Preload: the volume of blood in the ventricles before contraction. (2) Increasing the volume in the ventricles, consequently lengthening the myocardial fibers before contraction, should result in improved stroke volume (Frank–Starling law). The newborn infant is capable of increasing stroke volume provided there is no increase in systemic vascular resistance (SVR) or rapid rise in aortic pressure. Given the neonate’s smaller contractile mass, less compliant myocardium, and normal maximization of myocardial fiber length, volume infusions are likely to increase aortic pressure and afterload, resulting in a decline in stroke volume (Wernovsky and Gruber, 2005). b. Contractility: speed of ventricular contraction. (2) Ventricular contractility cannot be directly measured. Measurements of cardiac shortening fractions and ejection fraction provide an indirect assessment of contractility. (3) Contractility in neonates is influenced by the following: (b) Factors that decrease contractility: (i) Acidosis, which impairs myocardial response to catecholamines. (ii) Hypoxia. (iii) Electrolyte disturbances. (iv) Hypoglycemia. c. Afterload: the resistance to blood leaving the ventricle. (2) The neonate’s myocardium is very sensitive to increased afterload; with small increases in afterload, stroke volume can fall significantly. (3) Afterload can be reduced by intravenous (IV) infusion of vasodilators (e.g., nitroglycerin, amrinone, and nitroprusside). Dobutamine can be used to decrease PVR (Young and Mangum, 2008). 4. Concepts of blood flow. a. Flow is directly proportional to pressure/resistance. b. Blood flow will always take the path of least resistance. c. If heart action (pressure) remains unchanged but vasoconstriction or dilation or obstruction to flow (resistance) changes, flow will change (i.e., cardiac output will vary). d. PVR starts to fall by almost 80% after delivery, resulting in a dramatic increase in pulmonary blood flow and subsequent increase in systolic blood pressure (Blackburn, 2013). This normal decline is influenced by prematurity, low birth weight, and hypoxia episodes. For a list of abbreviations used for cardiac defects, see Table 28-3. TABLE 28-3 Abbreviations Used for Cardiac Defects A. Incidence: 3 to 9 per 1000 live births (Miller et al., 2011); the incidence in premature infants is 2 to 3 times that of term infants (Altman, 2013). 1. 85% of cases are due to multifactorial causes. 2. 10% to 12% are due to chromosomal factors. 3. 1% to 2% are due to genetic factors. 4. 1% to 2% are due to exposure to maternal or environmental teratogens (Lott, 2014). 5. 14.5% to 66% are in conjunction with other structural anomalies. B. Genetic factors: Chromosomal abnormalities: overall incidence of CHD in patients with a chromosome aberration is 30%. Isolated cardiac defects result from multiple genes and their interaction with factors in the environment. Advances in technology have identified genetic factors for many of these defects. 1. Incidence of CHD varies with chromosomal abnormality. Syndromes with an entire extra chromosome or syndromes with a missing chromosome are detected with normal chromosomal studies (van der Born et al., 2011): a. Trisomy 21: approximately 45%. b. Trisomy 18: 90% to 100%. c. Trisomy 13: 90%. d. Turner’s syndrome: 25% to 45% (45,X0 karyotype or mosaic 46,XX). 2. Syndromes requiring fluorescence in-situ hybridization (FISH) analysis include those with mosaicism and those with deletions in a chromosome. a. Chromosome deletion syndromes 18, 13, 5, and 4: 25% to 50%. b. Mosaic Turner’s syndrome: X and Y chromosome. FISH probe needed to rule out Y chromosome (Bishara and Clericuzio, 2008). 3. Microdeletions that are detected through FISH: a. 22q11 deletion syndrome: 80% to 85%. b. Williams syndrome: 50% to 70% (deletion of elastin gene at 7q11.23). c. Alagille syndrome: 90% (JAGI deletion on 20p12). d. Noonan’s syndrome: greater than 50% (12q24.1, PTPN11 gene). e. DiGeorge deletion: greater than 50% (22q11.2) (Hartman and Medoff-Cooper, 2012). 4. Single-mutant-gene syndromes: 1% to 2% of all cardiovascular abnormalities in newborn infants. Both autosomal dominant and recessive syndromes have been associated with CHD. According to Bishara and Clericuzio (2008), the tests for these syndromes include gene mutation analysis via array comparative genomic hybridization (aCGH). This type of testing can detect smaller imbalances in the genome, allowing for detection of mutations, insertions, deletions, and imprinting abnormalities. a. Holt–Oram syndrome: greater than 85% to 95% (TBX5 gene found at 12q24.1). b. X-linked situs inversus Xq25, ZIC3 gene. c. Kartagener’s syndrome (Fleiner, 2006): 100% (DNAI1 on 9p21-p13, DNAH5 on 5p15-p14). d. Beckwith–Wiedemann syndrome: 10% to 35%, imprinting defects of KIP2/LIT1 and paternal IGF2 genes at 11p15.5 (Jonas and Demmer, 2007). 5. Multifactorial: approximately 90% due to genetic predisposition coupled with a causative factor. a. Cornelia de Lange’s syndrome: approximately 30%. b. VACTERL (vertebral anomalies, anal atresia/stenosis, cardiac defects [50%], tracheoesophageal fistula, radial defects, limb anomalies) (Wechsler and Wernovsky, 2004). c. CHARGE (coloboma, heart defects [50% to 70%], choanal atresia, restriction of growth and development, genital anomalies, ear anomalies) (Wechsler and Wernovsky, 2004). 6. Genetic counseling is done to evaluate cause and recurrence risk. The risk of having another child with a cardiac lesion is 3% to 4% if the cause is unknown (Beck and Hudgins, 2003). However, this risk may be as much as 13% higher in specific syndromes. a. DiGeorge syndrome: Chromosomal microarray analysis should be recommended as first-line test if the infant has developmental disabilities (Hartman, et al., 2011). (1) 7% of parents carry the deletion. (2) Recurrence risk is up to 50% (Bishara and Clericuzio, 2008). (3) Prenatal diagnosis in future pregnancies via chorionic villus sampling (CVS) or amniocentesis, with FISH requested specifically. b. Trisomies 13, 18, and 21 all carry recurrence risk of 1%, and prenatal diagnosis in future pregnancies is done using CVS or amniocentesis. The recurrence risk for trisomy 21 increases after maternal age 40. C. Environmental factors and teratogens can include fetal exposure to drug, chemical, infectious, or physical agents; maternal disease or altered metabolic state with the period of greatest risk at 14 to 50 days of fetal life (Lott, 2014). 1. Cardiac teratogenesis is associated with maternal ingestion of the following: a. Thalidomide: dramatically illustrated the extraordinarily deleterious effects of prescribed drug on a developing fetus; included PDA, ASD, VSD, and pulmonary stenosis (Smithells and Newman, 1992). b. Anticonvulsants: 2% to 3% of infants exposed to anticonvulsants will have associated congenital cardiac defects such as pulmonary stenosis, coarctation of the aorta, and PDA. Glickstein (2007) reported the following defects with use of specific anticonvulsants in pregnancy: (2) Carbamazepine (Tegretol): VSD, TOF. (3) Valproic acid: VSD, coarctation of the aorta, interrupted aortic arch, TOF, HLHS, aortic stenosis, ASD, pulmonary atresia. (4) Trimethadione (fetal trimethadione syndrome): VSD, TGV, TOF, HLHS, double-outlet right ventricle, pulmonary atresia, ASD, aortic stenosis, pulmonic stenosis. (5) Pentobarbital: no confirmed specific embryopathy but may potentiate the effects of other drugs taken concurrently. c. Anticoagulants. (2) Heparin: because of its larger molecular weight, does not cross the placenta. d. Antineoplastic medications. (2) In general, the disorders for which antineoplastic agents are used are serious enough to preclude pregnancy (Glickstein, 2007). e. Lithium: approximately 10% of infants exposed will have defects such as Ebstein’s anomaly, ASD, and tricuspid atresia. f. Retinoic acid: aortic arch abnormalities, other congenital heart malformations. g. Isotretinoin. h. Alcohol: fetal alcohol syndrome (FAS) is accompanied by a variety of cardiac lesions (e.g., VSD with or without subpulmonic and subaortic stenosis, ASD, coarctation of the aorta, TOF). i. Amphetamine: 5% to 10% will have congenital cardiac defects such as VSD, PDA, ASD, and TGV (Kensey et al, 2011). j. Selective serotonin reuptake inhibitors: increased risk of VSD if used in first trimester (Koren and Nordeng, 2012). k. Tobacco and smoking: correlation between first-trimester smoking and defects such as pulmonary valve stenosis, TGV, ASD, right ventricular outflow tract defects, and PDA in term newborns (Alverson et al., 2011). 2. Exposure to environmental hazards such as radiation, heat, and gases may produce teratogenic effects, but there are no specific associated cardiac malformations. 3. Assisted reproductive technology highly linked to cardiac defects such as outflow tract abnormalities, VSD, TGV, and ASD (Tararbit et al., 2011). D. Maternal disease and viral infections (Box 28-1). 2. Maternal lupus erythematosus may be linked to complete congenital heart block and dilated cardiomyopathy. 3. Rubella and cytomegalovirus produce clinically significant heart disease (Kensey et al, 2011). Disorders seen are PDA, pulmonary stenosis and branch pulmonary artery stenosis, VSD, and ASD. 4. Maternal obesity has been linked to congenital heart disease (Glickstein, 2007). 5. Maternal fever and influenza are linked to right ventricular outflow tract and atrioventricular defects (Oster et al., 2011). 2. Aortic stenosis. 3. TGV. 4. HLHS. B. Females. 2. PDA. b. Left-to-right shunts (e.g., VSD or atrioventricular canal). 2. Term. The majority of infants with CHF are term infants. B. Maternal history (see Box 28-1). 1. Infants of mothers with uncontrolled diabetes have an increased risk of: b. VSD. c. Cardiomyopathy. d. Complex congenital heart disease. 2. Lupus may cause congenital heart block and cardiomyopathy. 3. Viral and bacterial illnesses may directly and indirectly affect the fetal cardiovascular system. 4. Seizures or coagulation disorders can be linked to CHDs because the medications used may be teratogens. 5. Maternal congenital heart disease can lead to an increased risk of same defect in infant. 6. Maternal age greater than 35 years increases risk. 7. Alcohol consumption: increase in cardiac defects seen with FAS. C. Familial. 1. Incidence of CHDs increases by 3- to 4-fold when a first-order relative (parent or sibling) has CHD (Oyen et al., 2009; Park, 2008). Risk increases by 10-fold if two first-order relatives have congenital heart disease (Table 28-4). TABLE 28-4 Familial Recurrence Risks Data from Park, M.K.: Pediatric cardiology for practitioners (5th ed.). Philadelphia, 2008, Elsevier Mosby; Theorell, C.: Cardiovascular assessment of the newborn. Newborn and Infant Nursing Reviews, 2(2):111-127, 2002; Altman, C.A.: Congenital heart disease (CHD) in the newborn: Presentation and screening for CHD. UpToDate. 2013. Available at www.uptodate.com. HLHS, Hypoplastic left heart syndrome; TGA, transposition of the great arteries.
Cardiovascular Disorders
CARDIOVASCULAR EMBRYOLOGY AND ANATOMY
Cardiac Development
Age (Days)
Cardiovascular Morphogenesis
18
Angiogenic tissue appears
19 to 20
Endothelial heart tubes appear
21
Fusion of endothelial heart tubes
23 to 25
Differentiation of atria, ventricles, and bulbus cordis; heart begins to beat
25
Fusion of atria, dorsal mesocardium regresses, formation of bulboventricular loop
26 to 27
Circulation is established
32
Appearance of septum primum
33
Intraventricular septum begins to form
35
Foramen secundum present
36 to 37
Fusion of atrial endocardial cushions
43
Ridges form in the bulbus cordis
46
Closure of ventricular septum, formation of coronary arteries
47
Septum secundum appears
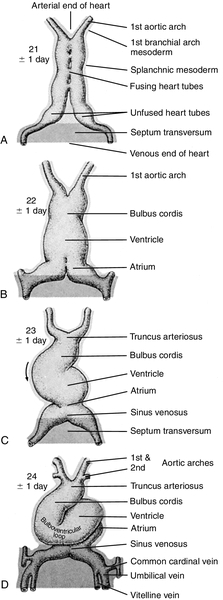
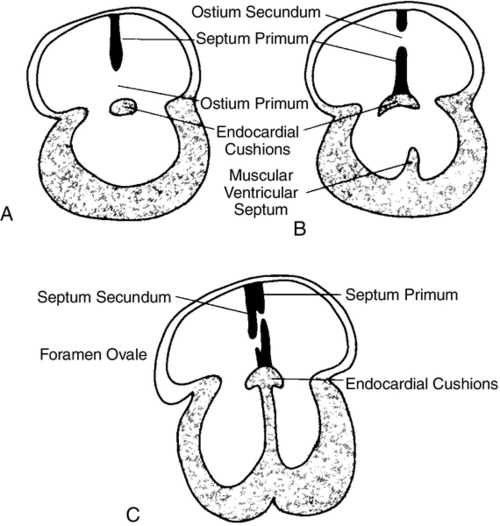
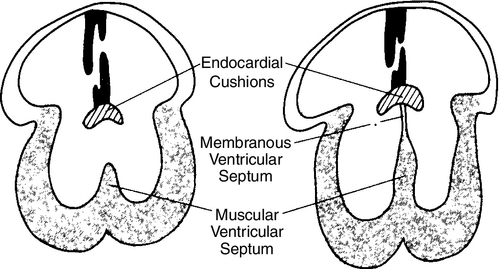
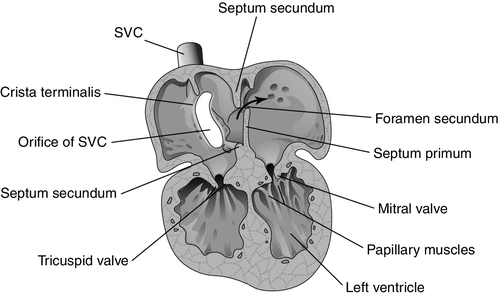
Great Vessel Development
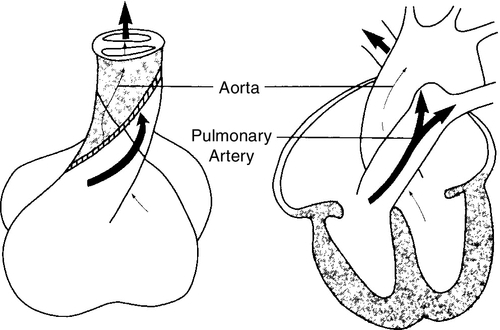
Embryonic Vessel
Results In
Truncus arteriosus
Aorta and pulmonary artery
First pair of aortic arches
Pieces remain as stapedial arteries
Second pair of aortic arches
Pieces remain as stapedial arteries
Third pair of aortic arches
Common carotid arteries and proximal portion of internal carotid arteries
Fourth pair of aortic arches
Right
Proximal portion of the right subclavian artery
Left
Aortic arch segment between left common carotid and left subclavian artery
Fifth pair of aortic arches
Regresses totally
Sixth pair of aortic arches
Left
Proximal portion of the left pulmonary artery and ductus arteriosus
Right
Proximal portion of the right pulmonary artery
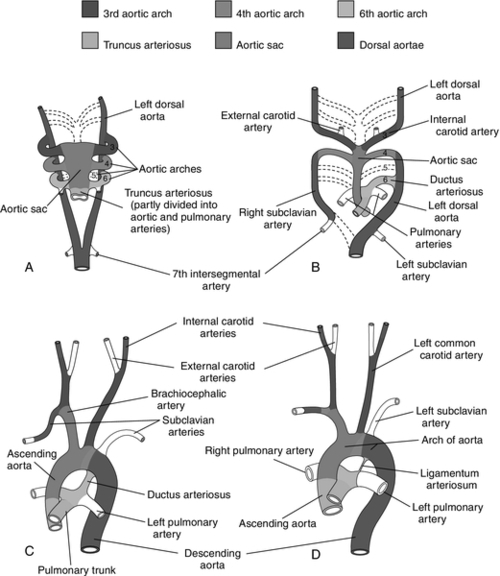
Circulatory Development
Cardiovascular Physiology
CONGENITAL HEART DEFECTS
AS
Aortic stenosis
ASD
Atrial septal defect
CoA
Coarctation of the aorta
HLHS
Hypoplastic left heart syndrome
IAA
Interrupted aortic arch
PA
Pulmonary atresia
PDA
Patent ductus arteriosus
PFO
Patent foramen ovale
PS
Pulmonary stenosis
TA
Truncus arteriosus
TAPVR
Total anomalous pulmonary venous return
TGV or TGA
Transposition of the great vessels or arteries
VSD
Ventricular septal defect
Occurrence
RISK ASSESSMENT AND APPROACH TO DIAGNOSIS OF CARDIAC DISEASE
Sex Preferences Associated With Cardiac Lesions
History (Kensey et al, 2011)
Cardiac Anomaly
Family Member with Congenital Heart Defect
Risk (%)
Overall
Parent
3.2
Grandparent
1.8
Aunt/Uncle/Cousin
1.1
PDA—patent ductus arteriosus
Mother
3.5 to 4
Father
2.5
Sibling
3
VSD—ventricular septal defect
Mother
6
Father
2
Sibling
3
ASD—atrial septal defect
Mother
4 to 4.5
Father
1.5
Sibling
2-3
AVC—atrioventricular canal
Mother
14
Father
1
Sibling
3-4
Tetralogy of Fallot
Mother
6 to 10
Father
1.5
Sibling
2.5
Aortic stenosis
Mother
13 to 18
Father
3
Sibling
2
Pulmonary stenosis
Mother
4 to 6.5
Father
2
Sibling
2
Coarctation of the aorta
Mother
4
Father
2
Sibling
2
Other defects (TGA, tricuspid atresia, truncus arteriosus, HLHS)
Sibling
1.4 (average)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


