The Child with a Condition of the Blood, Blood-Forming Organs, or Lymphatic System
Objectives
1. Define each key term listed.
2. Summarize the components of blood.
3. Recall normal blood values of infants and children.
4. List two laboratory procedures commonly performed on children with blood disorders.
5. Compare and contrast four manifestations of bleeding into the skin.
6. List the symptoms, prevention, and treatment of iron-deficiency anemia.
7. Recommend four food sources of iron for a child with iron-deficiency anemia.
8. Examine the pathophysiology and the signs and symptoms of sickle cell disease.
9. Describe four types of sickle cell crises.
10. Devise a nursing care plan for a child with sickle cell disease.
12. Review the effects of severe anemia on the heart.
13. Recall the pathophysiology and the signs and symptoms of hemophilia A and hemophilia B.
14. Identify the nursing interventions necessary to prevent hemarthrosis in a child with hemophilia.
15. Plan the nursing care of a child with leukemia.
16. Review the nursing care of a child receiving a blood transfusion.
17. Discuss the effects of chronic illness on the growth and development of children.
18. Recall the stages of dying.
19. Contrast age-appropriate responses to a sibling’s death and the nursing interventions required.
20. Formulate techniques the nurse can use to facilitate the grieving process.
21. Discuss the nurse’s role in helping families to deal with the death of a child.
Key Terms
alopecia (ăl-ō-PĒ-shă, p. 636)
Christmas disease (p. 633)
ecchymosis (ĕk-ĭ-MŌ-sĭs, p. 634)
erythropoietin (ĕ-rĭth-rō-POI-ă-tĭn, p. 625)
hemarthrosis (hē-măhr-THRŌ-sĭs, p. 633)
hematoma (hē-mă-TŌ-mă, p. 634)
hematopoiesis (hē-mă-tō-poi-Ē-sĭs, p. 625)
hemosiderosis (hē-mō-sĭd-ŭr-Ō-sĭs, p. 631)
lymphadenopathy (lĭm-făd-ĕn-ŎP-ă-thē, p. 626)
oncologists (p. 635)
petechiae (pĕ-TĒ-kē-ă, p. 626)
purpura (PŬR-pyŭ-ră, p. 627)
respite care (RĔS-pĭt kār, p. 641)
sickle cell crises (p. 629)
Sickledex (p. 631)
splenomegaly (splĕ-nō-MĒG-ă-lē, p. 626)
 http://evolve.elsevier.com/Leifer
http://evolve.elsevier.com/Leifer
The blood and blood-forming organs make up the hematological system. Blood is vital to all body functions. Blood dyscrasias or disorders occur when blood components fail to form correctly or when blood values exceed or fail to meet normal standards.
Plasma and blood cells are formed at about the second week of gestation, primarily in the yolk sac. Later, blood forms in the spleen, liver, thymus, lymph system, and bone marrow. In the fetus, blood is formed primarily in the liver until the last trimester of pregnancy. During childhood, the red blood cells (RBCs) are formed in the marrow of the long bones (such as the tibia and femur); by adolescence, hematopoiesis (blood formation) takes place in the marrow of the ribs, sternum, vertebrae, pelvis, skull, clavicle, and scapulae. The rate of RBC production is regulated by erythropoietin. This substance is produced by the liver of the fetus, but at birth the kidneys take over erythropoietin production.
The lymphatic system includes lymphocytes, lymphatic vessels, lymph nodes, the spleen, the tonsils, the adenoids, and the thymus gland. The lymphatic system drains regions of the body to lymph nodes, where infectious organisms are destroyed and antibody production is stimulated. Lymph nodes are not palpable in the newborn, but the cervical, axillary, and inguinal nodes may be palpable by childhood. Lymphadenopathy is an enlargement of lymph nodes that is indicative of infection or disease. Figure 27-1 summarizes some of the differences between the child’s and the adult’s lymphatic systems.
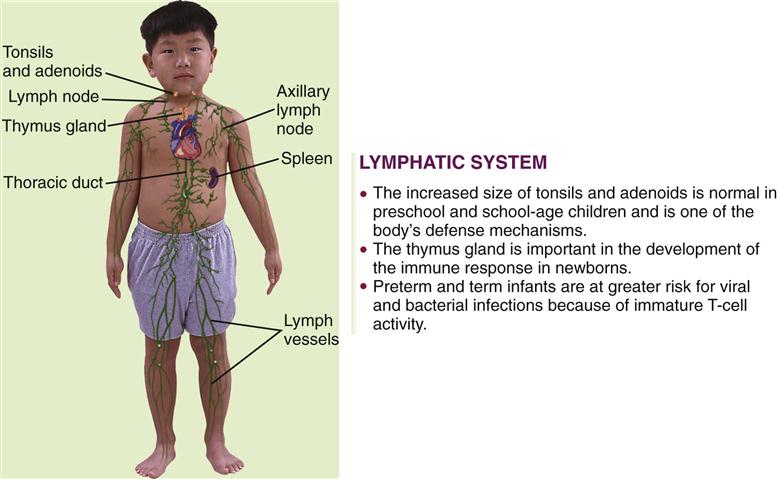
Figure 27-2 depicts the main types of blood cells in the circulating blood. Circulating blood consists of two portions: plasma and formed elements. The formed elements are erythrocytes (red blood cells), leukocytes (white blood cells [WBCs]), and thrombocytes (platelets). Erythrocytes primarily transport oxygen and carbon dioxide to and from the lungs and tissues. Leukocytes act as the body’s defense against infections. Thrombocytes, along with portions of blood plasma, are involved with blood coagulation. In the young child, every available space in the bone marrow is involved with blood formation.
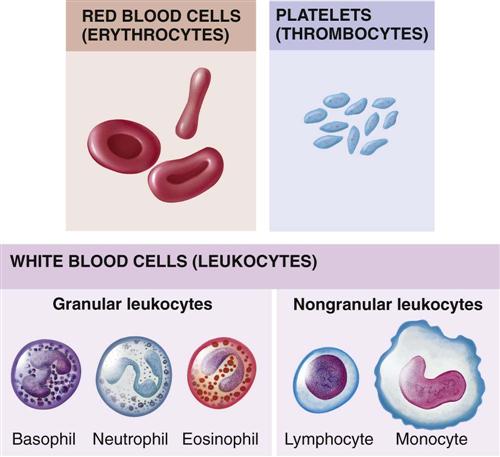
Lymphocytes, unlike other WBCs, are produced in the lymphoid tissues of the body. They travel in the circulation but are more commonly found in the lymph tissue. They are released into the body to fight infection and provide immunity. Their numbers greatly increase in chronic inflammatory conditions. The spleen is the largest organ of the lymphatic system. One of the main functions of the spleen is to bring blood into contact with lymphocytes. Aside from trauma and rupture, the most commonly seen pathological condition of the spleen is enlargement. This is termed splenomegaly. The spleen enlarges during infections, congenital and acquired hemolytic anemias, and liver malfunction.
Bone marrow aspiration is a procedure helpful in determining disorders of the blood. Numerous types of blood counts are used as well. Many are specific to a particular disease. The skin is sometimes an indicator of certain conditions of the blood. Petechiae (pinpoint hemorrhagic spots) and purpura (large petechiae) are often seen and should alert the nurse to the possibility of blood dyscrasias. The physician examines the liver and spleen by palpation and percussion to determine if they are enlarged.
Anemias
Anemia can result from many different underlying causes. A reduction in the amount of circulating hemoglobin reduces the oxygen-carrying ability of the blood. A hemoglobin level below 8 g/dL results in an increased cardiac output and a shunting of blood from the periphery of the body to the vital organs. Pallor, weakness, tachypnea, shortness of breath, and congestive heart failure can result.
Iron-Deficiency Anemia
Pathophysiology.
The most common nutritional deficiency of children in the United States today is anemia caused by insufficient amounts of iron in the body. The incidence is highest during infancy and adolescence—two rapid growth periods. Anemia (an, “without,” and emia, “blood”) is a condition in which there is a reduction in the amount and size of the RBCs or in the amount of hemoglobin, or both. Iron-deficiency anemia may be caused by severe hemorrhage, the child’s inability to absorb the iron received, excessive growth requirements, or an inadequate diet. Researchers have also found that feeding whole cow’s milk to infants can precipitate gastrointestinal bleeding, resulting in anemia.
Prevention of iron-deficiency anemia begins with good prenatal care to ensure that the mother has a suitable intake of iron during pregnancy. During the first few months after birth, the newborn relies on iron that was stored in the system during fetal life. Preterm infants may be deprived of a sufficient supply, because iron is obtained late in the prenatal period. In addition, the iron stores of low-birth-weight infants and infants from multiple births are relatively small.
The highest incidence of iron-deficiency anemia occurs from the ninth to the twenty-fourth month. During this rapid growth period, the infant outgrows the limited iron reserve that was in the body; in addition, iron-fortified formula and infant cereals may have been eliminated from the diet. Poorly planned meals or feeding problems also contribute to this deficiency. The mother may sometimes rely too heavily on bottle feedings to avoid conflict at meals. Unfortunately, milk contains very little iron. Instead, the amounts of solid food should be increased and the amount of milk decreased. Boiled egg yolk, liver, leafy green vegetables, Cream of Wheat, dried fruits (apricots, peaches, prunes, raisins), dry beans, crushed nuts, and whole-grain bread are good sources of iron. Iron-fortified cereals eaten out of the box provide a nutritious snack. Unfortunately, not all the iron found in a food source is absorbed by the body. The bioavailability of iron in vegetables is less than that in meat.
Manifestations.
The symptoms of iron-deficiency anemia are pallor, irritability, anorexia, and a decrease in activity. Many infants are overweight because of excessive consumption of milk (so-called “milk babies”). Blood tests for anemia may include RBC count, hemoglobin and hematocrit levels, and determination of morphological cell changes and iron concentration. The stool may be tested for occult blood. A dietary history is also obtained. Sometimes a slight heart murmur is heard. The spleen may be enlarged.
Untreated iron-deficiency anemias progress slowly, and in severe cases the heart muscle becomes too weak to function. If this happens, heart failure follows. Children with long-standing anemia may also show growth retardation and cognitive changes. Screening procedures are suggested at 9 and 24 months for full-term infants and earlier for low-birth-weight infants.
Treatment.
Iron-deficiency anemia responds well to treatment. Iron, usually ferrous sulfate, is given orally two or three times a day between meals. Vitamin C aids in the absorption of iron; therefore giving juice when administering iron is suggested. Liquid preparations are taken through a straw to prevent temporary discoloration of the teeth. (Some iron preparations without this disadvantage are available.) The toddler needs solid foods that are rich sources of iron. An iron-dextran mixture (Imferon) given intramuscularly is also highly effective. It must be injected deep in a large muscle using the Z-track technique to minimize staining and tissue irritation.
 Nursing Tip
Nursing Tip
Oral iron supplements should not be given with milk or milk products because milk interferes with iron absorption.
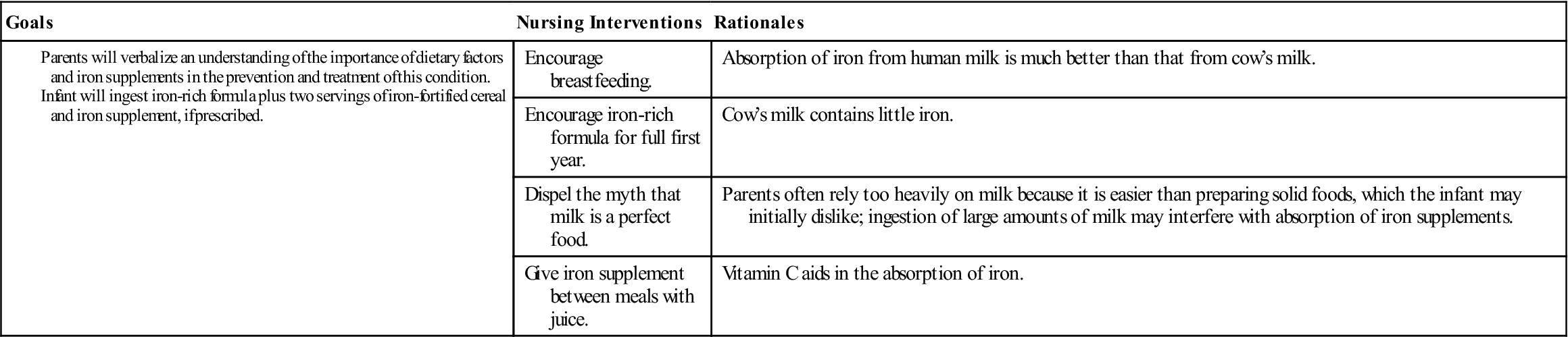
Parent Education.
Parents need explicit instructions on proper foods for the infant. The nurse stresses the importance of breastfeeding for the first 6 months and the use of iron-fortified formula throughout the first year of life (the absorption of iron from human milk is much better than from cow’s milk). The amount of milk consumed during the day and night is determined. Solid food intake is reviewed, and specific iron-enriched nutrients are suggested. The nurse considers financial, ethnic, and family preferences in teaching plans. Behavior concerns at mealtime may also have to be addressed.
The stools of infants who are taking oral iron supplements are tarry green. An absence of this finding may indicate poor parental compliance with therapy. Oral iron preparations are not to be given with milk, which interferes with absorption. To increase absorption, these preparations should be given between meals, when digestive acid concentration is highest. It is important to emphasize that both dietary changes and supplemental iron therapy are necessary to eradicate iron-deficiency anemia. Good dietary practices must be lifelong to maintain good health. Parents are encouraged to return for periodic evaluation of the child’s blood status. Nursing Care Plan 27-1 specifies interventions for the child with iron-deficiency anemia.
 Medication Safety Alert!
Medication Safety Alert!
Prevent iron poisoning in children by keeping preparations well out of reach. Educate parents about this hazard.
27-1
 Nursing Care Plan
Nursing Care Plan
The Child with Iron-Deficiency Anemia
Patient Data
A parent brings a 10-month-old boy to the clinic. The child is pale and listless. Laboratory results show iron-deficiency anemia. The parent states that the child loves to drink milk.
Selected Nursing Diagnosis
Deficient parental knowledge related to cause and treatment of anemia
Selected Nursing Diagnosis
Imbalanced nutrition: less than body requirements, related to iron-poor diet
| Goals | Nursing Interventions | Rationales |
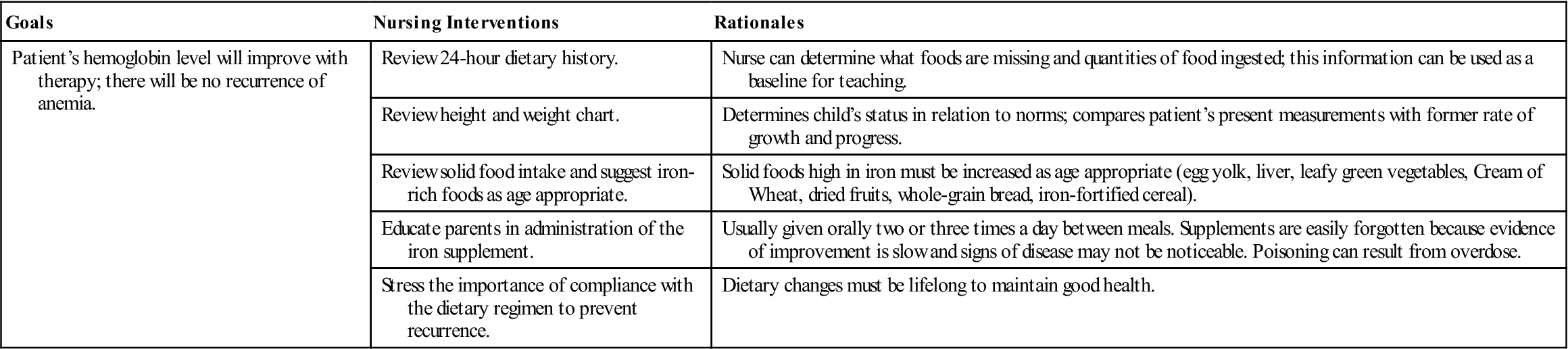
| Patient’s hemoglobin level will improve with therapy; there will be no recurrence of anemia. | Review 24-hour dietary history. | Nurse can determine what foods are missing and quantities of food ingested; this information can be used as a baseline for teaching. |
| Review height and weight chart. | Determines child’s status in relation to norms; compares patient’s present measurements with former rate of growth and progress. | |
| Review solid food intake and suggest iron-rich foods as age appropriate. | Solid foods high in iron must be increased as age appropriate (egg yolk, liver, leafy green vegetables, Cream of Wheat, dried fruits, whole-grain bread, iron-fortified cereal). | |
| Educate parents in administration of the iron supplement. | Usually given orally two or three times a day between meals. Supplements are easily forgotten because evidence of improvement is slow and signs of disease may not be noticeable. Poisoning can result from overdose. | |
| Stress the importance of compliance with the dietary regimen to prevent recurrence. | Dietary changes must be lifelong to maintain good health. |
Critical Thinking Question
Sickle Cell Disease
Pathophysiology.
Sickle cell disease is an inherited defect in the formation of hemoglobin. It occurs mainly in people of African descent, but it is also carried by some persons of Mediterranean descent, such as some Arabs, Greeks, Maltese, Sicilians, as well as those originating from other Mediterranean areas. Many researchers believe that the gene for sickle cell disease developed in these populations as protection against malaria. Sickling (clumping) caused by decreased blood oxygen levels may be triggered by dehydration, infection, physical or emotional stress, or exposure to cold. Laboratory examination of the affected child’s blood shows that the RBC has changed its shape to resemble that of a sickle blade, from which the name of the disorder is derived (Figure 27-3).
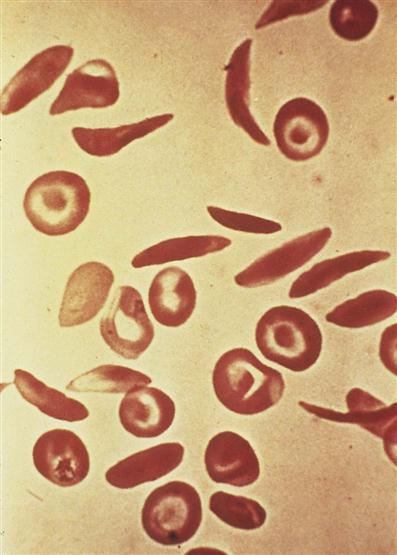
Sickle cells contain an abnormal form of hemoglobin termed hemoglobin S (the sickling type). The membranes of these cells are fragile and easily destroyed. Their crescent shape makes it difficult for them to pass through the capillaries, causing a pileup of cells in the small vessels. This clumping may lead to a thrombosis (clot) and cause an obstruction. Infarcts, or areas of dead tissue, may result when the tissue is denied proper blood supply. These generally develop in the spleen but may also be seen in other areas of the body, such as the brain, heart, lungs, gastrointestinal tract, kidneys, and bones. The patient feels acute pain in the affected area.
There are two types of sickle cell disease: an asymptomatic (a, “without,” and symptoma, “symptom”) version (sickle cell trait) and much more severe forms that necessitate intermittent hospitalization (sickle cell anemia).
Sickle cell trait.
This form of the disease occurs in about 10% of the African American population in the United States. The blood of the patient contains a mixture of normal (hemoglobin A) and sickle (hemoglobin S) hemoglobins. The proportions of hemoglobin S are low because the disease is inherited from only one parent. The physician can distinguish sickle cell trait from the more severe disease by electrophoresis study of the patient’s RBCs and hemoglobin. Sickling is more rapid and extreme with sickle cell disease. In sickle cell trait the hemoglobin and RBC counts are normal.
Sickle cell trait does not develop into sickle cell disease. Although there is no need to treat the patient with sickle cell trait, the patient is a carrier, and genetic counseling is important.
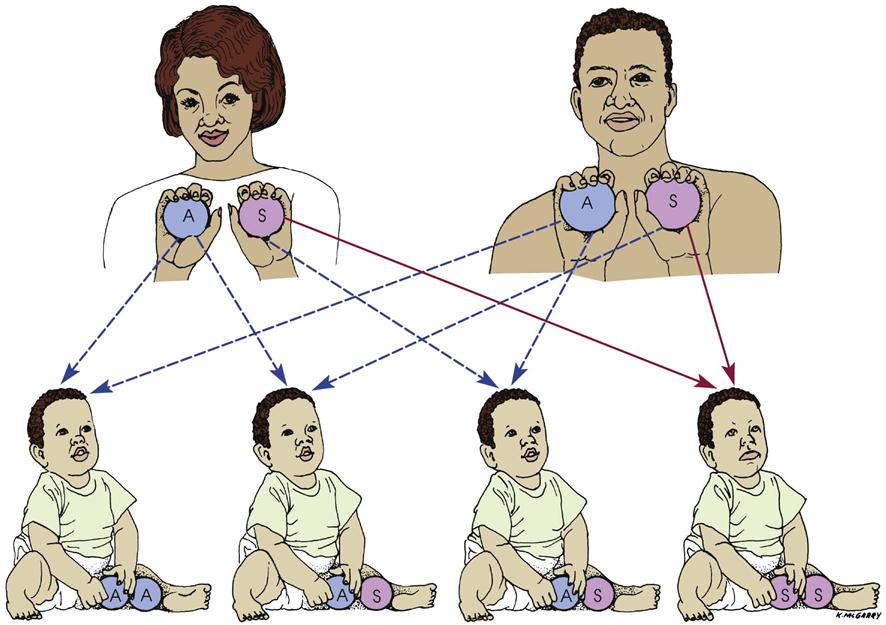
Sickle cell anemia.
This severe form of sickle cell disease results when the abnormality is inherited from both parents (Figure 27-4). Each offspring has a one in four chance of inheriting the disease (which is not the same as one in four children inheriting it). In general, the clinical symptoms do not appear until the last part of the first year of life. There may be an unusual swelling of the fingers and toes. The symptoms of sickle cell disease are caused by enlarging bone marrow sites that impair circulation to the bone and the abnormal sickle cell shape that causes clumping, obstruction in the vessel, and ischemia to the organ that the vessel supplies.
There is chronic anemia. The hemoglobin level ranges from 6 to 9 g/dL or lower. The child is pale, tires easily, and has little appetite. These manifestations of anemia are complicated by characteristic episodes called sickle cell crises, which are painful and can be fatal. Specific types of crises have been defined. They differ in their pathological causes and manifestations and may necessitate somewhat different treatments (Table 27-1). Unfortunately, in some cases the sickle cell crisis is the first obvious manifestation of the condition. The patient appears acutely ill. There is severe abdominal pain. Muscle spasms, leg pain, or painful swollen joints may be seen. Fever, vomiting, hematuria, convulsions, stiff neck, coma, or paralysis can result, depending on the organs involved. Children with sickle cell disease have a risk for stroke as a complication of a vasoocclusive sickle cell crisis.
Table 27-1
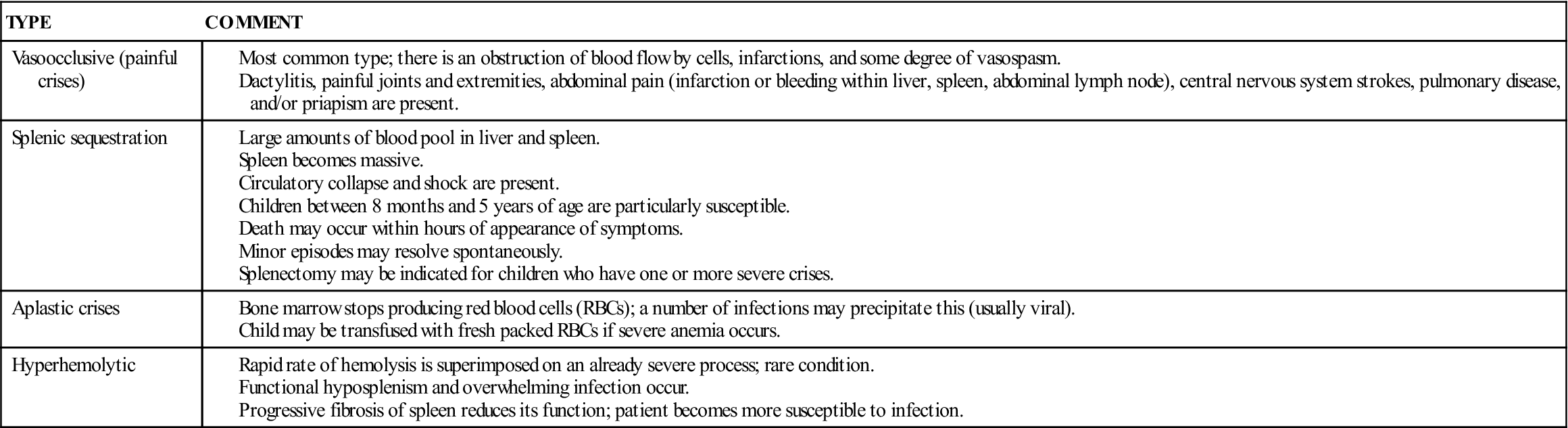
Data from Jackson, P., Vessey, J., & Schapiro, N (2009). Primary care of the child with a chronic condition (5th ed.). Philadelphia: Saunders; Kliegman, R., Behrman, R., Jenson, H., & Stanton, B. (2007). Nelson textbook of pediatrics (18th ed.). Philadelphia: Saunders.
The sickle cell crises recur periodically throughout childhood; however, they tend to decrease with age. Patients should be kept in good health between episodes. Immunizations of these children are particularly important, including those for Haemophilus influenzae, hepatitis A, and hepatitis B as well as the pneumococcal vaccine. Patients should refrain from becoming overly tired. They also should avoid situations such as flying in an unpressurized airplane or exercising at a high altitude, because oxygen concentrations in their blood are already reduced. Extra stress and exposure to cold may lower resistance, causing additional problems. Overheating, which can lead to dehydration, is also to be avoided. Oral intake of iron is of no value.
 Nursing Tip
Nursing Tip
During sickle cell crises, anticipate the child’s need for tissue oxygenation, hydration, rest, protection from infection, pain control, blood transfusion, and emotional support for this life-threatening illness.
Diagnosis.
Sickle cell disease can be detected before birth by chorionic villi sampling. Early diagnosis by mandatory screening of all newborns in the United States allows early detection before symptoms occur. The sickling test (Sickledex) is commonly used for screening. Hemoglobin electrophoresis (“fingerprinting”) is used when the result is positive. This procedure separates and records the various peptide patterns of the blood. It distinguishes between patients with the trait and those with the disease.
Treatment and Nursing Care.
When the infant or child is hospitalized during a crisis, the treatment is supportive and symptomatic. The patient is confined to bed. Analgesics are given to relieve pain. Children in severe pain may need a continuous intravenous (IV) infusion containing a narcotic. Meperidine (Demerol) is not recommended for children with sickle cell disease because of the risk of normeperidine-induced seizures (National Institutes of Health [NIH], 2002). A patient-controlled analgesia (PCA) pump enables children older than 7 years to maintain control and participate in care. Every effort is made to combat dehydration and acidosis. Small blood transfusions may be administered to increase the hemoglobin count, but the results are only temporary. Packed RBCs are often given for this purpose. An accurate record of intake and output is kept. The patient’s body position is changed frequently but gently. Hemopoietic stem cell transplantation may provide a cure in the near future.
Sickle cell disease may take a wide variety of courses. As always, the individual patient’s progress is followed. Sometimes it is difficult to distinguish between abdominal pain caused by a sickle cell crisis and abdominal pain caused by appendicitis. The nurse must remember that pain experienced by a child with sickle cell disease may also be caused by an unrelated condition.
Prevention of infection and prevention of dehydration are important goals in the care of a child with sickle cell disease. Pneumococcal and meningococcal vaccinations and annual influenza vaccination are recommended. Oral penicillin prophylaxis before any invasive treatment, including dental care, is also advised (NIH, 2002). Multiple transfusions of packed cells may be required to maintain adequate hemoglobin levels. The nurse should observe the child for reactions to the blood transfusion. Hemosiderosis (the deposit of iron into organs and tissues in the body) is a complication of this type of hemolytic disease. An exchange transfusion may reduce the number of circulating sickle cells and prevent complications such as thrombus formation and stroke. High-dose IV methylprednisolone may be used to decrease severe pain (Dover & Platt, 2003).
The main goals of therapy are to prevent sickling, dehydration, hypoxia, and infection, which can cause a sickle cell crisis. Erythropoietin and some chemotherapy regimens can increase the production of fetal hemoglobin and reduce complications. Bone marrow transplantation and gene replacement therapy may hold a promising cure (Jackson et al., 2009).
 Medication Safety Alert!
Medication Safety Alert!
Meperidine (Demerol) should not be used for pain control in children with sickle cell disease.
Surgery.
The use of splenectomy in children with sickle cell disease has been conservative. A recurrence of acute splenic sequestration becomes less likely after age 5 years. Routine splenectomy is not recommended because the spleen generally atrophies on its own because of fibrotic changes that take place in patients with sickle cell disease.
 Nursing Tip
Nursing Tip
It is essential to teach parents signs and symptoms of dehydration, hypoxia, and infection to prevent the occurrence of sickle cell crisis.
Thalassemia
Pathophysiology.
The thalassemias are a group of hereditary blood disorders in which the patient’s body cannot produce sufficient adult hemoglobin. The RBCs are abnormal in size and shape and are rapidly destroyed. This abnormality results in chronic anemia. The body attempts to compensate by producing large amounts of fetal hemoglobin. Thalassemias are caused by a deficiency in the normal synthesis of hemoglobin polypeptide chains. They are categorized according to the Greek letters designating the polypeptide chain affected as α-, β-, γ-, or δ-thalassemia.
The most common variety of thalassemia involves impaired production of beta chains and is known as β-thalassemia. This variety consists of two forms: thalassemia minor and thalassemia major. Thalassemia major is also called Cooley’s anemia. Thalassemia occurs mainly in persons of Mediterranean origin, such as Greeks, Syrians, and Italians, and their descendants elsewhere. The term is derived from the Greek thalassa, which means “sea.” Thalassemia can also occur from spontaneous mutations.
Thalassemia minor.
Thalassemia minor, which is also termed β-thalassemia trait, occurs when the child inherits a thalassemia gene from only one parent (heterozygous inheritance). It is associated with mild anemia. Hemoglobin concentration averages 2 to 3 g/dL, lower than normal age-related values. These patients are often misdiagnosed as having an iron-deficiency anemia. Symptoms are minimal. The patient is pale, and the spleen may be enlarged. The patient may lead a normal life, with the illness going undetected. This condition is of genetic importance, particularly if both parents are carriers of the trait. Prenatal blood samples can detect thalassemia major in such cases.
Thalassemia major (Cooley’s anemia).
When two thalassemia genes are inherited (homozygous inheritance), the child is born with a more serious form of the disease. A progressive, severe anemia becomes evident within the second 6 months of life.
The child is pale and hypoxic, has a poor appetite, and may have a fever. Jaundice, which at first is mild, progresses to a muddy bronze color resulting from hemosiderosis, a deposit of iron (released by blood cell destruction) into the tissues. The liver enlarges, and the spleen grows enormously. Abdominal distention is great, which causes pressure on the organs of the chest. Cardiac failure caused by the profound anemia is a constant threat. Bone marrow space enlarges to compensate for an increased production of blood cells. Hematopoietic (hema, “blood,” and poiesis, “to make”) defects and a massive expansion of the bone marrow in the face and skull result in changes in the facial contour that give the child a characteristic appearance (Figure 27-5). The teeth protrude because of an overgrowth of the upper jawbone; the bone becomes thin and is subject to pathological fracture.
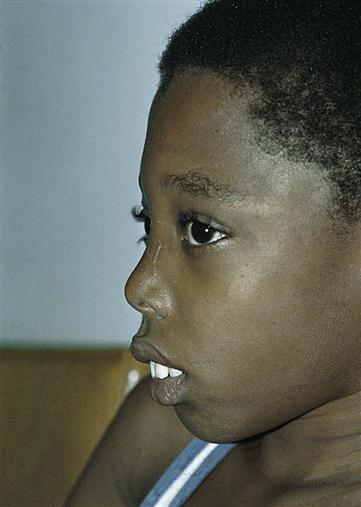
Diagnosis is aided by a family history of thalassemia, radiographic bone growth studies, and blood tests. Hemoglobin electrophoresis is helpful in diagnosing the type and severity of the various thalassemias. Prenatal screening and diagnosis are available, and genetic counseling is advised.
Treatment and Nursing Care.
The goals of care for children with thalassemia are to (1) maintain hemoglobin levels at 9.5 g/dL to prevent overgrowth of bone marrow and resultant deformities and (2) provide for growth and development and normal physical activity. Prevention or early treatment of infection is important. Some patients require splenectomy. Bone marrow transplantation holds promise for the future treatment of this condition (Jackson et al., 2009).
The mainstay of treatment for thalassemia major is frequent blood transfusions to maintain the hemoglobin level above 9.5 g/dL. As a result of repeated blood transfusions, excessive deposits of iron may be stored in the tissues. This is termed hemosiderosis and is seen especially in the spleen, liver, heart, pancreas, and lymph glands. Deferoxamine mesylate (Desferal), an iron-chelating agent, is given to counteract hemosiderosis. Severe splenomegaly may occur in some children. Splenectomy may make the patient more comfortable, increase the ability to move about, and allow for more normal growth. After surgery, these children are given prophylactic antibiotics to prevent infection.
Nursing measures adhere to the principles of long-term care. The observation of the patient during a blood transfusion is discussed on p. 637. Monitoring of vital signs is necessary to detect irregularities of the heart. Whenever possible, the same nurse cares for the patient during transfusions, blood tests, and other unpleasant procedures to provide security. Children are taught to regulate their activities according to their own tolerance.
The emotional health of the child and parents calls for special consideration by the nurse. Every attempt to ease the strain of this prolonged illness must be made. Home care arrangements can be provided through community agencies. The family can be referred to the Cooley’s Anemia Foundation for support and education. Older children need special support to accept changes in their body image caused by the disease. Suggestions applicable to the care of the chronically ill child are discussed throughout the text. Care of the dying child is discussed later in this chapter.
Bleeding Disorders
Hemophilia
Pathophysiology.
Hemophilia is one of the oldest hereditary diseases known to humanity. In this disorder the blood does not clot normally, and even the slightest injury can cause severe bleeding. It has been called the disease of kings because it has occurred in the children of several royal families in Russia and Europe. This congenital disorder is confined almost exclusively to males but is transmitted by symptom-free females.
Hemophilia is inherited as a sex-linked recessive trait. It is termed sex-linked because the defective gene is located on the X, or female, chromosome. Different combinations of genes account for the fact that some children inherit the disease, some become carriers, and others neither inherit nor carry the trait. New mutations do occur, and the reason for this is unclear. The sex of the fetus can be determined by amniocentesis. Fetal blood sampling detects hemophilia. Some carrier women can also be identified.
There are several types of hemophilia. More than 10 identified factors in blood are involved in the clotting mechanism. A deficiency in any one of the factors will interfere with normal blood clotting. The two most common types of hemophilia are hemophilia B, or Christmas disease (a factor IX deficiency), and hemophilia A (a deficiency in factor VIII). This discussion is limited to classic hemophilia, or hemophilia A, which accounts for approximately 84% of cases.
Hemophilia A is caused by a deficiency of coagulation factor VIII, or antihemophilic globulin (AHG). The severity of the disease depends on the level of factor VIII in the plasma of the patient’s blood. Some patients’ lives are endangered by minor injury, whereas a child with a mild case of hemophilia might just bruise a little more easily than the normal person. The degree of severity tends to remain constant within a given family. The aim of therapy is to increase the level of factor VIII high enough to ensure clotting. It is possible to determine the level of factor VIII in the blood by means of a test called the partial thromboplastin time (PTT), which can help to diagnose and assess the child’s condition. Prenatal diagnosis by amniocentesis is possible.
 Nursing Tip
Nursing Tip
A classic symptom of hemophilia is bleeding into the joints (hemarthrosis).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


