The Newborn at Risk
Acquired and Congenital Conditions
Objectives
2. Discuss the prenatal diagnosis of Down syndrome.
3. Recognize three genetic inborn errors of metabolism.
5. Describe common congenital anomalies.
6. Interpret signs associated with elevated bilirubin in the newborn.
7. Explain the nursing interventions used in phototherapy.
8. Articulate the principles of newborn resuscitation.
9. Outline the common respiratory problems in the newborn.
10. Characterize the effect of maternal diabetes on the newborn.
11. Outline six problems of infants born to mothers with diabetes mellitus.
12. Explain factors responsible for newborn sepsis, and state the nurse’s role in reducing the risks.
13. Discuss the nursing assessment that would lead the nurse to suspect newborn sepsis.
14. Identify the defects involved in the tetralogy of Fallot and common manifestations.
15. Compare the alteration of blood flow of cyanotic and noncyanotic congenital heart defects.
16. Explain the pathophysiology of noncyanotic congenital heart defects.
17. Describe care of the newborn who has neonatal abstinence syndrome.
Key Terms
galactosemia (gă-lăk-tō-SĒ-mē-ă, p. 323)
hemolysis (hē-MŎL-ĭ-sĭs, p. 329)
hyperglycemia (hĭ-pĕr-glĭ-SĒ-mē-ă, p. 336)
hypoglycemia (hĭ-pō-glĭ-SĒ-mē-ă, p. 332)
infants of diabetic mothers (IDMs) (p. 336)
kernicterus (kĕr-NĬK-tĕr-ŭs, p. 329)
macrosomia (măk-rō-SŌ-mē-ă, p. 336)
meconium aspiration syndrome (MAS) (mă-KŌ-nē-ŭm, p. 334)
neonatal sepsis (nē-ō-NĀ-tăl, p. 337)
phenylketonuria (PKU) (fĕn-ŭl-kē-tō-NŪ-rē-ă, p. 322)
phototherapy (p. 330)
 http://evolve.elsevier.com/Leifer/maternity
http://evolve.elsevier.com/Leifer/maternity
Genetics is the scientific study of the transmission of characteristics from parent to child (see Chapters 1 and 3). Genetic defects are common causes of acute and chronic conditions that manifest during fetal life; immediately after birth; or during childhood, adolescence, or adulthood. An understanding of human genetics, including genetic challenges, disorders, and technology, is an integral part of maternity nursing and medical obstetric practice.
Birth defects, abnormalities that are apparent at birth, occur in 3% to 4% of all live births. The rate is even higher if the defects that become evident later in life are counted. An abnormality of structure, function, or metabolism may result in a physical or mental disability, may shorten life, or may be fatal. Box 16-1 shows the system of classification of birth defects. Because these disorders include so many conditions, it is necessary to limit the number discussed in the chapter and to place others in relevant areas of the text (see the index for specific conditions). Defects that are manifested later in life are discussed in pediatric textbooks.
Defects present at birth often involve the skeletal system; limbs may be missing, malformed, or duplicated. Some abnormalities (e.g., congenital hip dysplasia) are more subtle, and the nurse must be alert to detect them. Inborn errors of metabolism include a number of inherited diseases that affect body chemistry. There may be an absence or a deficiency of a substance necessary for cell metabolism. The deficient substance is usually an enzyme. Almost any organ of the body may be damaged. Examples of inborn errors of metabolism include cystic fibrosis and phenylketonuria (PKU). In disorders of the blood, there is a reduced or missing blood component or an inability of a component to function adequately. Sickle cell disease, thalassemia, and hemophilia fall into this category. Chromosomal abnormalities number in the thousands. Most involve some type of mental retardation, and others are incompatible with life. The newborn with Turner’s syndrome or Klinefelter’s syndrome may have impaired physical growth and sexual development. Perinatal injuries have many causes and are seen in various forms, the most common of which is premature birth.
As the March of Dimes Foundation (2007) points out, “Few birth defects can be attributed to a single cause. The majority are thought to result from an interplay between environment and heredity, depending on inherited susceptibility, stage of pregnancy, and degree of environmental hazard.” Newborns with birth defects may need to remain in the neonatal unit for an extended time for intensive care and treatment. Screening tests are performed prenatally to detect many genetic defects and, in some cases, enable appropriate treatment before or immediately after birth (see Chapter 5).
Chromosomal Disorders
The human body is made up of 23 paired chromosomes, with one pair from the mother and one pair from the father. These chromosomes contain deoxyribonucleic acid (DNA) and other complex proteins. An abnormal chromosome number or arrangement can cause a congenital defect. Sometimes an extra chromosome is present or a chromosome is broken or missing. The resulting imbalance can provide the newborn with too little or too much genetic material. As the embryo grows, the scrambled genetic information may translate into various types of congenital defects.
Down Syndrome
Trisomy 21, also known as Down syndrome, is one of the most common chromosomal syndromes, occurring in 1 in 600 to 800 live births (Gabbe, Simpson, & Niebyl, 2007). It affects all races and economic levels equally. Down syndrome often results from having an extra chromosome (usually chromosome 21) and is called trisomy 21 because it was first identified with this chromosome. Occasionally, the extra chromosome 21 is attached to another chromosome in the egg or sperm, which can create a translocation (an alteration in location). The translocation in either parent greatly increases the chances of having another child with Down syndrome. Mothers older than 35 years are at the greatest risk of having a Down syndrome newborn.
The American Academy of Pediatrics recommends screening of all pregnant women for Down syndrome in the first trimester of pregnancy. First trimester screening includes ultrasound assessment of the thickness of the fetal nuchal fold (called nuchal translucency). An ultrasound demonstrating absence of the nasal bone (Cicero, Avgidou, Rembouskos, et al., 2006) and a second trimester “quad test” involving blood testing of alpha-fetoprotein (AFP), human chorionic gonadotropin (hCG), unconjugated estriol (UE), and inhibin A (a placental hormone) are additional screening tests for Down syndrome. A low AFP or a high hCG and inhibin A, with a low UE, may indicate a high risk for Down syndrome for the developing fetus. A test of pregnancy-associated plasma protein A (PAPP-A) may also indicate a risk for Down syndrome. Positive tests in the first or second trimester may indicate a need for amniocentesis to confirm the diagnosis.
Characteristics of the Down syndrome newborn are most noted in the craniofacial features (Figure 16-1). The eyes have an upward slant because of an epicanthal fold, speckles known as Brushfield’s spots are seen in the iris, the nose is small with a wide nasal bridge, and the ears are low set. The tongue appears large and protrudes from the newborn’s mouth, fingers are short and broad, often there is an unusually wide space between the first two toes, and a single palmar crease (simian crease) may be present. Relatively common internal anomalies in trisomy 21 newborns include heart defects and duodenal atresia. Mental retardation is exhibited, with the mean IQ of approximately 50 (range: 25 to 70). Females are fertile; however, it is rare for males to be fertile.
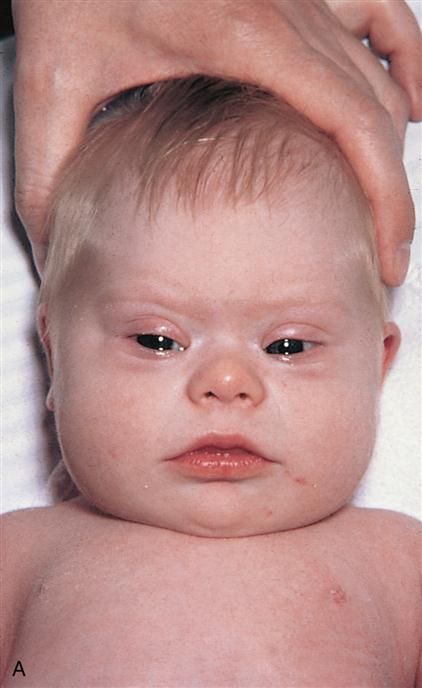
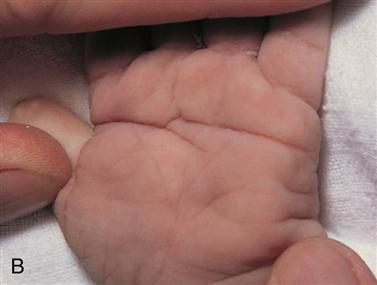
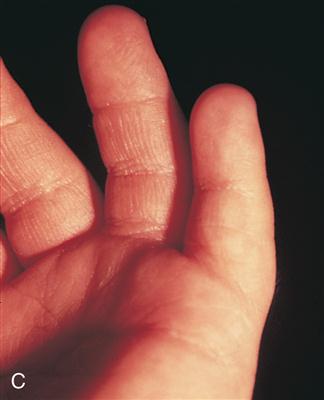
At birth, these newborns are usually hypotonic (limp and flaccid) and may have feeding difficulties. They also have increased susceptibility to respiratory tract infections. Parents need guidance in feeding and preventing infections and encouragement to stimulate their newborns developmentally. Children with Down syndrome are usually very affectionate. Parents should be encouraged to join a support group for parents of children with Down syndrome. Developmental disabilities, including mental retardation, are common, with hearing and speech difficulties that complicate efforts at education. Cardiac, orthopedic, and thyroid dysfunction are also common. Alzheimer’s disease often develops early in the third decade of life, and an altered immune response increases susceptibility to respiratory and dental infection.
Inborn Errors of Metabolism
Inborn errors of metabolism do not always manifest symptoms at birth. Screening tests for specific errors of metabolism are required by law in most states. It is important for early identification so that treatment can be started as soon as possible to minimize effects on the newborn.
Phenylketonuria
Phenylketonuria (PKU) is an autosomal recessive inherited inborn error of phenylalanine metabolism that occurs in 1 in 15,000 live births (Trahms, 2008). It is caused by the faulty metabolism of phenylalanine, an amino acid that is essential to life and found in all protein foods. The hepatic enzyme phenylalanine hydrolase, which is required to convert phenylalanine into tyrosine, is missing. As a result, when the newborn ingests protein (found in milk and all protein foods), phenylketones accumulate in the blood and can rise as high as 20 times the normal level. Its byproduct, phenylpyruvic acid, appears in the urine within the first weeks of life. These phenylketones accumulate in the brain and cause irreversible brain damage, resulting in severe mental retardation. Early detection and treatment are essential because by the time the urine test is positive, brain damage has already occurred. The newborn appears normal at birth but begins to show delayed development at approximately 4 to 6 months of age. The newborn may show evidence of failure to thrive or have eczema or other skin conditions. Characteristically, these newborns have an unusual musty odor to their body and in their urine.
A diagnosis is made by the Guthrie test. Blood is obtained from a simple heel prick, and a few drops of capillary blood are placed on a filter paper and mailed to the laboratory for screening. It is recommended that the blood be obtained after 48 to 72 hours of life, preferably after ingestion of proteins, to reduce the possibility of false results. All states in the United States require that the test be performed in all newborns before they leave the nursery, but, because of early discharge, the test is often repeated after discharge. The newborn can be tested at home by a public health nurse or at the physician’s office or clinic. Confirmation of the diagnosis requires quantitative elevations of phenylalanine compound in both blood and urine. The nurse must stress to the mother the importance of the return visit of the newborn to the physician or clinic for the repeat testing.
Since the development of newborn screening for PKU, several women who had been diagnosed with PKU in the newborn period and then were treated with a phenylalanine-restricted diet have grown up not suffering the damages of untreated PKU. However, newborns born to women with PKU who do not follow the restricted diet during pregnancy have teratogenic effects if high concentrations of phenylalanine are circulating in the mother’s blood. Congenital heart defects, microcephaly, and mental retardation are the most commonly seen effects. Therefore, for these women, following dietary restrictions from conception (especially during embryonic development) is critical.
The newborn diagnosed with PKU is fed a special formula (Lofenalac) that has had the phenylalanine reduced or removed. Phenyl-Free is given to children, and Phenex-2 is given to adolescents. The goals of the diet are to provide enough essential proteins to support growth and development while maintaining phenylalanine blood levels between 2 and 10 mg/dL. A phenylalanine level less than 2 mg/dL may result in growth retardation, and a level greater than 10 mg/dL can result in significant brain damage. Levels are monitored throughout childhood.
A dietitian should be consulted for parental guidance and support in maintaining the dietary regimen. This attention is especially needed for the school-age child and the adolescent. The intake of most meat, dairy products, and diet drinks needs to be restricted, and protein intake is restricted to that required for basic growth. The milk substitute can be flavored with a fruit powder or a chocolate substitute, which can increase the child’s compliance. Aspartame, a sugar substitute, must be avoided. Genetic counseling is important for future family planning.
Galactosemia
Galactosemia is an inborn error of metabolism that occurs in 1 in 53,000 live births (Tarini & Freed, 2007). The newborn has a deficiency of the enzyme necessary to convert galactose to glucose, resulting in an increased amount of galactose in the blood (galactosemia), liver, brain, kidney, and urine (galactosuria). An early diagnosis is important so that a milk substitute can be prescribed because galactose is present in milk. Galactosemia can be detected by measuring blood levels of galactose, and screening of all newborns is performed in most states across the United States. Failure to thrive, cataracts, jaundice, cirrhosis of the liver, sepsis, and mental retardation are manifestations of untreated cases. Therapy consists of eliminating galactose from the diet and providing lactose-free formula, such as Nutramigen. Because breast milk contains lactose, breastfeeding must be discontinued. Medications that contain lactose fillers as inactive ingredients also must be avoided. Parents and children often experience frustration and anxiety and must be educated and supported in following this dietary program.
Hypothyroidism
Congenital hypothyroidism is the result of an inborn error of metabolism caused by a maternal iodine deficiency or the use of antithyroid drugs by the mother. It occurs 1 in 4000 live births (Palma-Sisto, 2004). Thyroxine (T4) is measured from a drop of blood obtained from heel stick at 2 to 5 days of age. If not treated with thyroid replacement, the infant may develop hypothermia, poor feeding, lethargy, jaundice, and cretinism. The infant has a large protruding tongue, thick lips, and a generally dull appearance.
An important note is that one blood sample can be used to test all three metabolic disorders: PKU, galactosemia, and hypothyroidism. The screening test for hypothyroidism is mandated in all states across the United States and is often performed before discharge from the nursery. Early diagnosis and treatment are essential to maintain normal physical and mental growth and development.
Maple Syrup Urine Disease
Maple syrup urine disease is a disorder of amino acid metabolism in which the amino acids leucine, isoleucine, and valine cannot be metabolized because of missing enzymes. Elevated levels of leucine can cause cerebral edema and central nervous system (CNS) symptoms such as seizures. Body fluids have a sweet odor, similar to maple syrup. Some states require routine screening of all newborns for this condition (see Chapter 9). Nursing responsibilities include educating the family concerning strict dietary and exercise limitations throughout the infant’s lifetime.
Common Congenital Anomalies
Although congenital anomalies are generally treated in the pediatric setting, they are usually identified soon after birth. The family of the newborn with the disorder may have been aware of the condition through prenatal testing, or they may be surprised by the birth of a newborn who is not completely normal. The family will need a great deal of support and sensitivity from the health care providers. Some congenital anomalies are noted in Table 16-1. The nursing student is urged to read a pediatric textbook for more detailed information concerning the management of congenital anomalies in the newborn.
Table 16-1
| Anomaly | Treatment and Nursing Care |
| Gastrointestinal | |
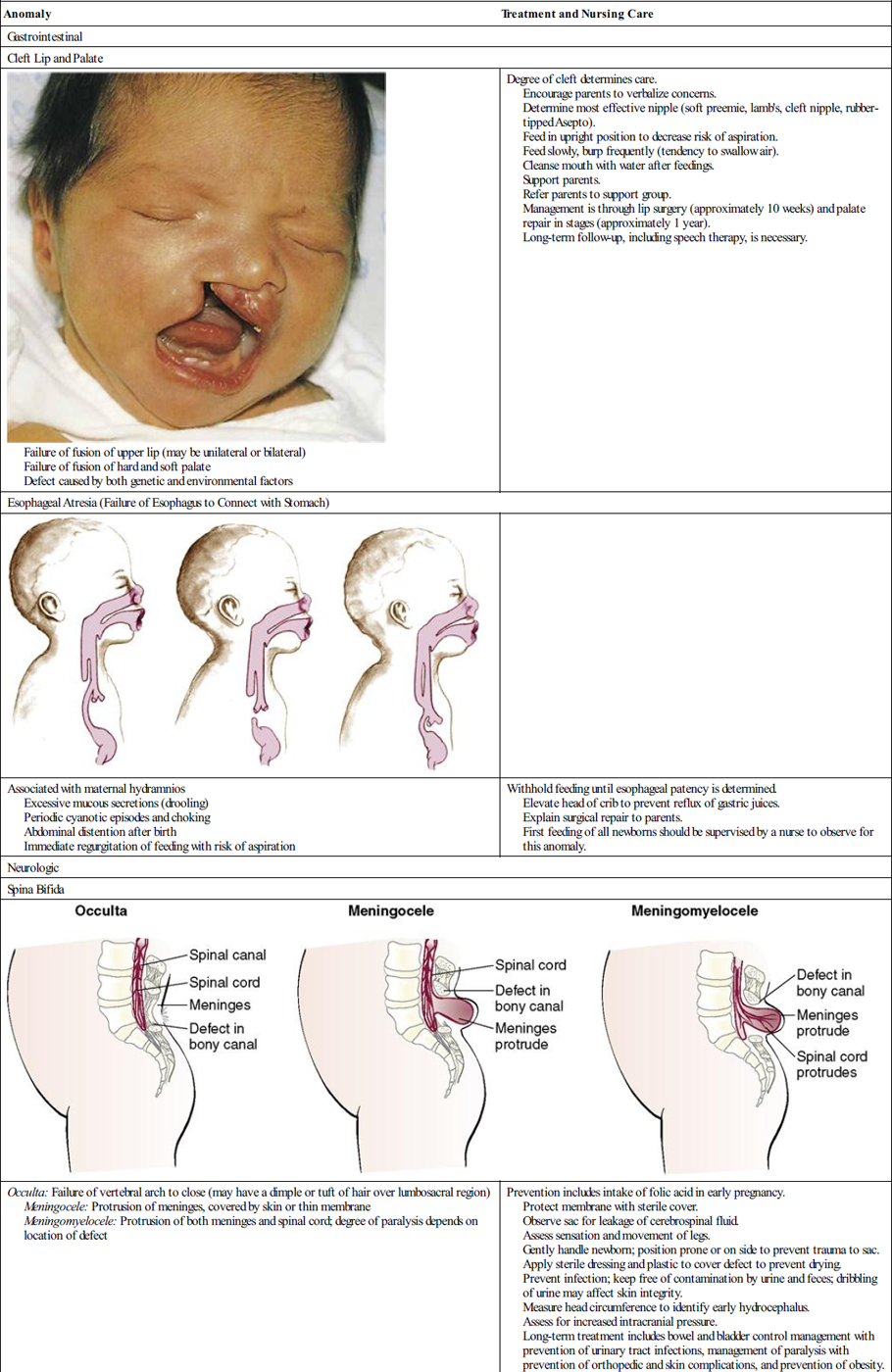
| Cleft Lip and Palate | |
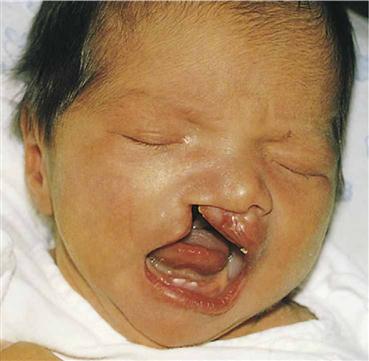 Failure of fusion of upper lip (may be unilateral or bilateral) Failure of fusion of hard and soft palate Defect caused by both genetic and environmental factors | Degree of cleft determines care. Encourage parents to verbalize concerns. Determine most effective nipple (soft preemie, lamb’s, cleft nipple, rubber-tipped Asepto). Feed in upright position to decrease risk of aspiration. Feed slowly, burp frequently (tendency to swallow air). Cleanse mouth with water after feedings. Support parents. Refer parents to support group. Management is through lip surgery (approximately 10 weeks) and palate repair in stages (approximately 1 year). Long-term follow-up, including speech therapy, is necessary. |
| Esophageal Atresia (Failure of Esophagus to Connect with Stomach) | |
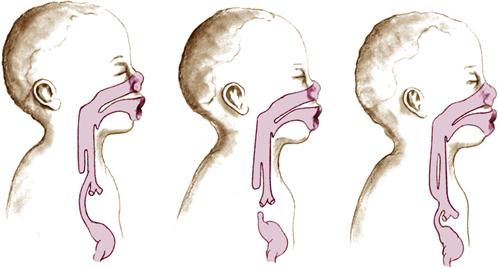 | |
| Associated with maternal hydramnios Excessive mucous secretions (drooling) Periodic cyanotic episodes and choking Abdominal distention after birth Immediate regurgitation of feeding with risk of aspiration | Withhold feeding until esophageal patency is determined. Elevate head of crib to prevent reflux of gastric juices. Explain surgical repair to parents. First feeding of all newborns should be supervised by a nurse to observe for this anomaly. |
| Neurologic | |
| Spina Bifida | |
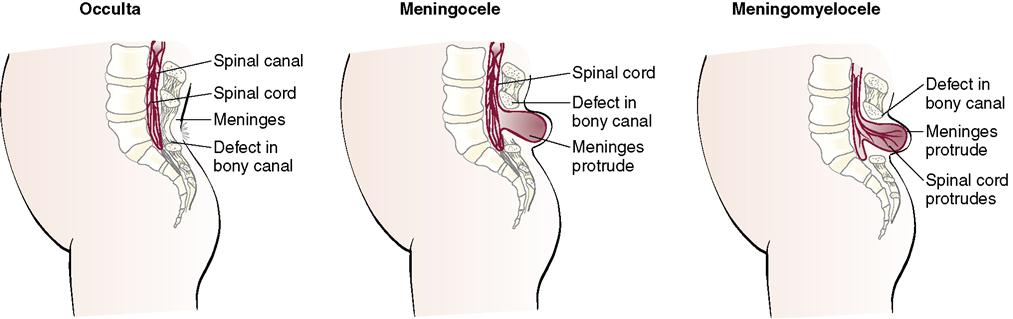 | |
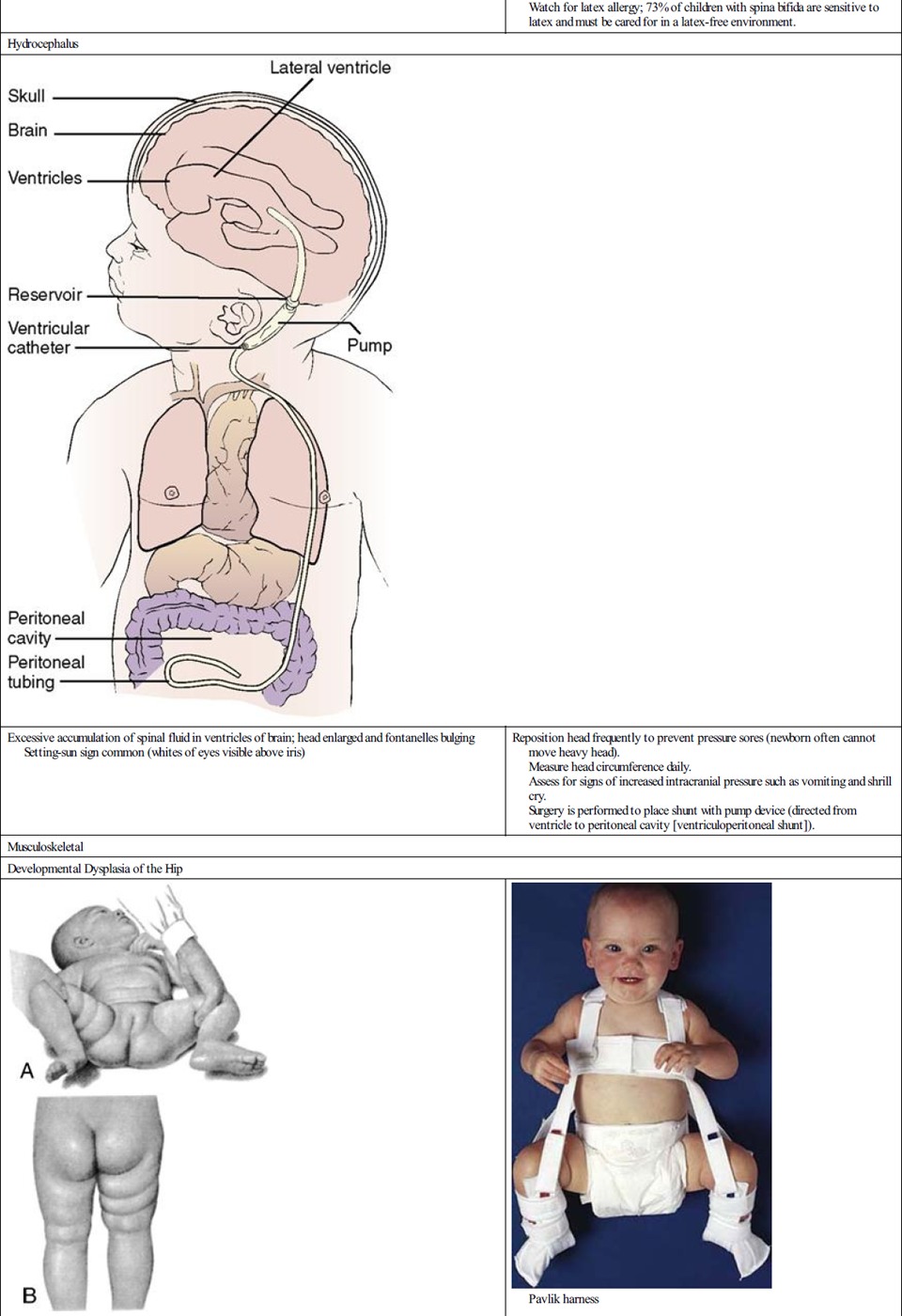
| Occulta: Failure of vertebral arch to close (may have a dimple or tuft of hair over lumbosacral region) Meningocele: Protrusion of meninges, covered by skin or thin membrane Meningomyelocele: Protrusion of both meninges and spinal cord; degree of paralysis depends on location of defect | Prevention includes intake of folic acid in early pregnancy. Protect membrane with sterile cover. Observe sac for leakage of cerebrospinal fluid. Assess sensation and movement of legs. Gently handle newborn; position prone or on side to prevent trauma to sac. Apply sterile dressing and plastic to cover defect to prevent drying. Prevent infection; keep free of contamination by urine and feces; dribbling of urine may affect skin integrity. Measure head circumference to identify early hydrocephalus. Assess for increased intracranial pressure. Long-term treatment includes bowel and bladder control management with prevention of urinary tract infections, management of paralysis with prevention of orthopedic and skin complications, and prevention of obesity. Watch for latex allergy; 73% of children with spina bifida are sensitive to latex and must be cared for in a latex-free environment. |
| Hydrocephalus | |
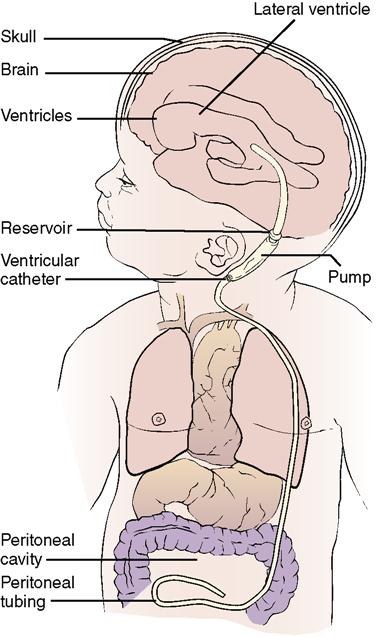 | |
| Excessive accumulation of spinal fluid in ventricles of brain; head enlarged and fontanelles bulging Setting-sun sign common (whites of eyes visible above iris) | Reposition head frequently to prevent pressure sores (newborn often cannot move heavy head). Measure head circumference daily. Assess for signs of increased intracranial pressure such as vomiting and shrill cry. Surgery is performed to place shunt with pump device (directed from ventricle to peritoneal cavity [ventriculoperitoneal shunt]). |
| Musculoskeletal | |
| Developmental Dysplasia of the Hip | |
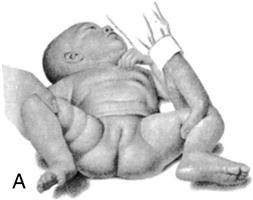 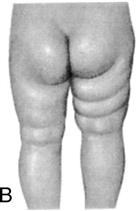 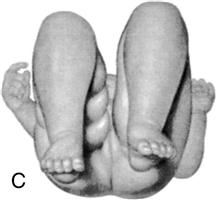 | 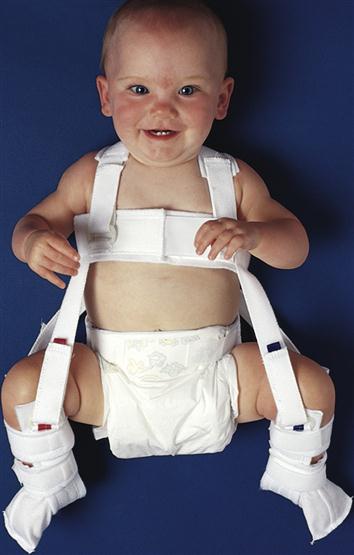 Pavlik harness |
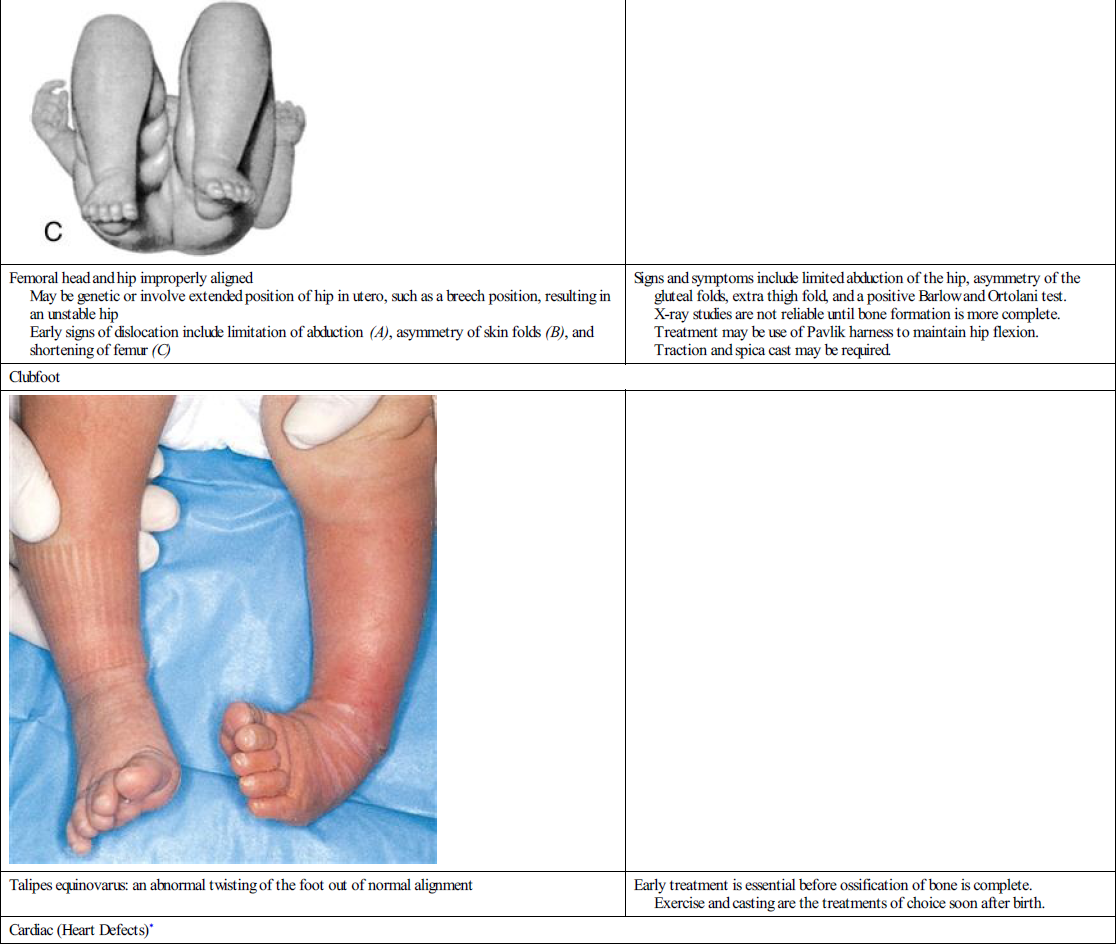
| Femoral head and hip improperly aligned May be genetic or involve extended position of hip in utero, such as a breech position, resulting in an unstable hip Early signs of dislocation include limitation of abduction (A), asymmetry of skin folds (B), and shortening of femur (C) | Signs and symptoms include limited abduction of the hip, asymmetry of the gluteal folds, extra thigh fold, and a positive Barlow and Ortolani test. X-ray studies are not reliable until bone formation is more complete. Treatment may be use of Pavlik harness to maintain hip flexion. Traction and spica cast may be required. |
| Clubfoot | |
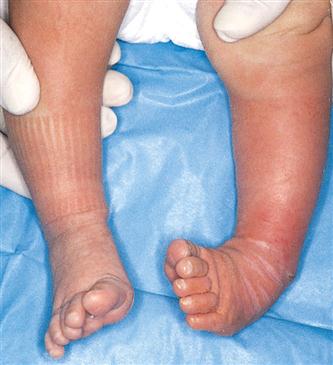 | |
| Talipes equinovarus: an abnormal twisting of the foot out of normal alignment | Early treatment is essential before ossification of bone is complete. Exercise and casting are the treatments of choice soon after birth. |
| Cardiac (Heart Defects)* | |
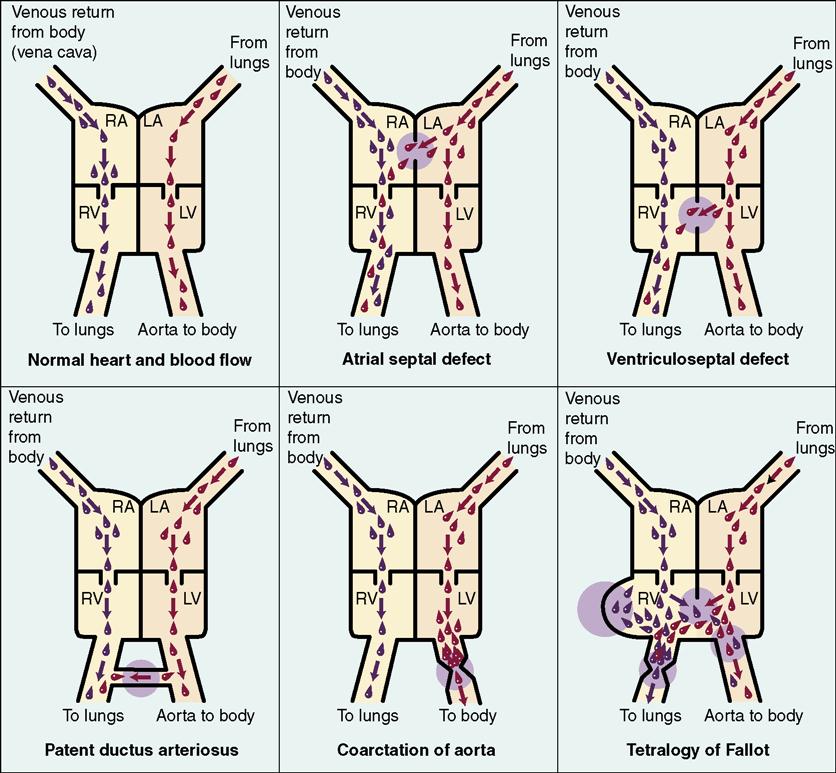 | |
| Patent Ductus Arteriosus | |
| Noncyanotic heart defect Failure of the ductus arteriosus, connecting the pulmonary artery and the aorta, to close after birth Cyanosis does not occur because blood recirculates to the lung and is fully oxygenated when it flows to general circulation | Heart defects may or may not be identified immediately. Newborn with heart defect may exhibit murmurs, abnormal heart rate or rhythm, breathlessness, and fatigue while feeding. Surgery may be postponed until newborn is physiologically stable. |
| Tetralogy of Fallot | |
| Cyanotic heart defect Involves four characteristic defects: a ventricular septal defect, aorta positioned over the ventricular septal defect, stenosis of the pulmonary valve, and hypertrophy of the left ventricle Cyanosis results from venous blood from the right ventricle flowing through the septal defect and directly into the overriding aorta; blood flow to the lungs is decreased because of the narrowed pulmonary valve; cyanosis occurs because unoxygenated blood reaches the general circulation | Monitor closely; observe for respiratory difficulties, cyanosis, tachycardia, tachypnea, diaphoresis. Conserve newborn’s energy to reduce workload on heart. Gavage feedings or oral feedings with special nipple may be given. Elevate newborn’s head and shoulders to improve respirations and reduce cardiac workload. Prevent infection. Place in knee-chest position for respiratory distress during “tet” attack. Management includes corrective surgery. |
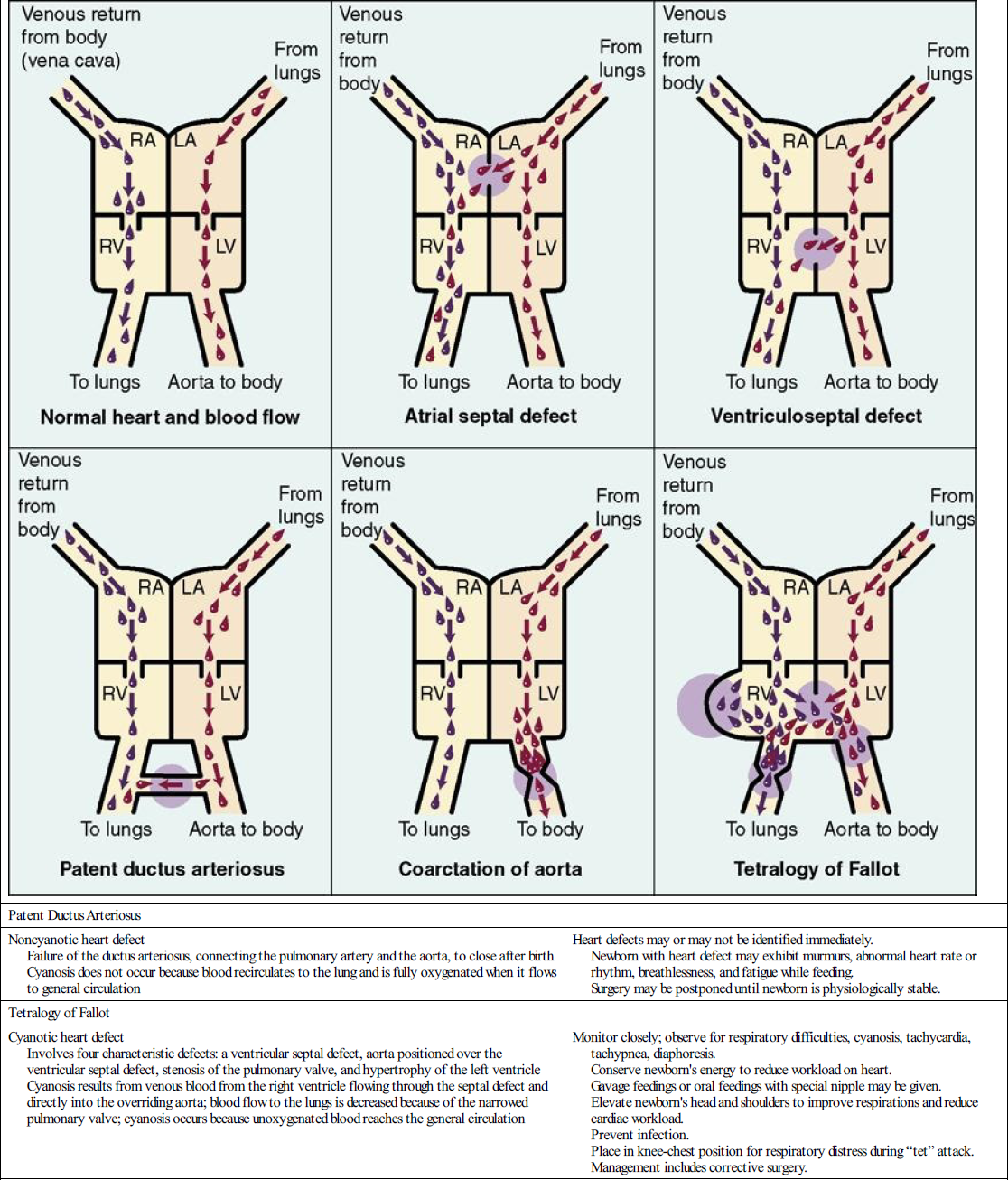
LA, left aorta; LV, left ventricle; RA, right aorta; RV, right ventricle.
*For other common congenital heart defects, students should consult their pediatric textbooks.
Common Acquired Disorders
Hyperbilirubinemia (Physiologic Jaundice)
Hyperbilirubinemia is defined as an abnormally high level of bilirubin in the blood. This condition occurs when the normal pathways of bilirubin metabolism and excretion in the newborn are altered because of excess products (from normal hemolysis), liver immaturity, delayed feeding (which prevents development of intestinal flora), trauma, or cold stress. An increase in bilirubin also can occur as a result of cephalhematoma, extensive bruising, infections, and acidosis that causes a decrease in liver function. Maternal use of sulfa or salicylates can interfere with conjugation of bilirubin. The result is clinical jaundice (icterus neonatorum), which is seen in approximately 60% of term newborns; it is even more common in preterm newborns, typically occurring after the third day of life. Total serum bilirubin levels greater than 12 mg/dL in the term newborn usually indicate hyperbilirubinemia.
The newborn is born with an excessive amount of red blood cells (RBCs) and, at birth, begins to destroy the RBCs he or she no longer needs. The infant does not need the excess RBCs because he or she is now in an atmosphere of higher oxygen concentration than was available in utero. Bilirubin inside the RBC is released into the bloodstream when the RBC is destroyed. The bilirubin combines with albumin in the blood and is transported to the liver. Under the influence of the enzyme glucuronyl transferase, the bilirubin is conjugated into a water-soluble form and excreted subsequently into the small intestine; most of it is excreted from the body in the feces. Some bilirubin is converted to unconjugated bilirubin, is reabsorbed, and is recirculated back into the blood. The amount of bilirubin in the blood is described in milligrams of bilirubin per deciliter (mg/dL). When the bilirubin accumulates in the blood, it contributes to a condition called icterus neonatorum, or physiologic jaundice. The skin and whites of the eyes assume a yellow-orange cast. The higher the bilirubin level the deeper the jaundice. An increase of more than 5 mg/dL in 24 hours or a bilirubin blood level of 12.9 mg/dL or more requires investigation and intervention.
The bilirubin level in newborns typically peaks between 3 and 5 days of age. Therefore, the early discharge of newborns requires follow-up observation within a few days. Conjugation of the bilirubin can be inhibited by a lack of bacteria in the intestines or a low level of glucuronyl transferase enzyme. The immature liver may also be slow to take up the bilirubin that flows to it. For these reasons, the liver may not be able to “clear” the bilirubin from the blood and excrete it from the body at a rapid rate. High levels of bilirubin in the blood can stain the basal nuclei of the brain, causing long-term neurologic problems. This condition is called kernicterus.
Because bilirubin conjugated by the liver is excreted by the body via the intestinal tract, the stimulation of meconium stool passage is an important part of the care plan. The initiation of early feedings enhances the passage of meconium and therefore plays an essential role in the management of hyperbilirubinemia. Colostrum, in breast milk, has a natural laxative effect, and therefore breastfeeding at least 10 times a day is recommended for the neonate. Glucose water supplements should be avoided because little bilirubin is excreted by the kidneys and decreased caloric intake is associated with decreased passage of stool, allowing for the reabsorption of bilirubin before it can be excreted. Although there is a factor in human milk that may increase reabsorption of bilirubin from the intestines, this rarely causes a significant rise in serum bilirubin. Some physicians may prefer to feed the infant bottled formula for a 12- to 24-hour period to avoid this small increase. The mother should be encouraged to pump milk from the breast during this short period to enable easy return to breastfeeding.
Assessment and Management of Physiologic Jaundice
All newborns with visible jaundice and all infants under 35 weeks’ gestation should have serum bilirubin levels drawn (Rennie, 2010). The visual blanch test is used to help distinguish jaundice from normal skin color. Pressure is applied with a finger over a bony area on the newborn, such as the nose, forehead, or sternum, for several seconds to empty the capillaries in that area. A yellow tinge in the blanched area indicates jaundice. When pressure is released, the capillaries refill. The conjunctivae of the eyes and the buccal mucosa can also be visually assessed to detect jaundice. Assessing for jaundice should be done under natural light because reflections of wall color can influence the appearance of skin color (Skill 16-1). The cephalocaudal (head-to-toe) pattern of circulation results in a head-to-toe progression of jaundice in the newborn so that jaundice affecting the head and upper body may be a reflection of a lower serum bilirubin level than in infants who evidence jaundice of the chest and lower body. Serum bilirubin levels are taken whenever the jaundice levels appear before 24 hours of age. A bilirubin threshold table is available as a guideline in the management of infants with hyperbilirubinemia (Rennie, 2010).
Noninvasive Methods of Bilirubin Measurement
The trauma, cost, and inconvenience of obtaining a blood sample for serum bilirubin levels can be saved by measuring the transcutaneous bilirubin (TcB). Hand-held electronic devices such as the BiliCheck (Respironics) measure TcB levels. TcB monitoring is a screening tool. Those infants with TcB bilirubin levels above 14.6 mg/dL should have serum bilirubin levels drawn and referred for further follow-up.
Hyperbilirubinemia (Pathologic Jaundice)
A major cause of hyperbilirubinemia is hemolytic disease, in which there is an excessive breakdown of RBCs of the newborn as a result of maternal antibodies passing through the placenta to the fetus in the uterus. Isoimmune hemolytic disease, also known as erythroblastosis fetalis, occurs when an Rh-negative mother is pregnant with an Rh-positive fetus and transplacental passage of maternal antibodies occurs. When maternal antibodies enter the fetal circulation, they destroy the fetal RBCs. The fetal system responds by increasing the RBC production with a marked increase in immature RBCs (erythroblasts). A high level of maternal antibodies may have developed during a previous pregnancy, abortion, amniocentesis, or abruptio placentae. A previous blood transfusion with Rh-positive blood would also cause the development of maternal antibodies. At birth, the newborn with hemolytic disease caused by Rh incompatibility has a positive direct Coombs’ test, which reveals the presence of antibody-coated (sensitized) Rh-positive RBCs in the newborn. The indirect Coombs’ test measures the amount of Rh-positive antibodies in the mother’s blood. If the mother prophylactically receives Rho(D) immune globulin (RhoGAM), maternal development of antibodies to Rh-positive blood greatly decreases (Figure 16-2). Pathologic jaundice, such as that caused by Rh incompatibility, is evident before the third day of life because the hemolysis begins before birth.

Pathologic Jaundice
In pathologic jaundice of erythroblastosis (Rh hemolytic disease), jaundice is evident before the third day of life. Jaundice that is first evident after the third day of life is usually caused by physiologic icterus neonatorum.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


