The Newborn with a Perinatal Injury or Congenital Malformation
Objectives
1. Define each key term listed.
2. List and define the more common disorders of the newborn.
3. Describe the classifications of birth defects.
4. Outline the nursing care for the newborn with hydrocephalus.
5. Describe the symptoms of increased intracranial pressure.
6. Discuss the prevention of neural tube anomalies.
7. Outline the preoperative and postoperative nursing care of a newborn with spina bifida cystica.
8. Differentiate between cleft lip and cleft palate.
9. Discuss the dietary needs of a newborn with phenylketonuria.
10. Discuss the early signs of developmental hip dysplasia.
11. Discuss the care of the newborn with Down syndrome.
12. Outline the causes and treatment of hemolytic disease of the newborn (erythroblastosis fetalis).
13. Devise a plan of care for a newborn receiving phototherapy.
14. Describe home phototherapy.
15. Discuss the assessment and nursing care of a newborn with macrosomia.
Key Terms
birth defects (p. 321)
cheiloplasty (KĪ-lō-plăs-tē, p. 328)
cleft lip (p. 327)
cleft palate (p. 328)
clubfoot (p. 329)
congenital malformations (p. 322)
erythroblastosis fetalis (ĕ-rĭth-rō-blăs-TŌ-sĭs fĕ-TĂL-ĭs, p. 337)
habilitation (p. 326)
hydrocephalus (hī-drō-SĔF-ă-lăs, p. 322)
hyperbilirubinemia (hī-pŭr-bĭl-ē-rū-bĭ-NĒ-mē-ă, p. 339)
kernicterus (p. 339)
macrosomia (măk-rō-SŌ-mē-ă, p. 344)
meconium aspiration syndrome (MAS) (p. 344)
meningocele (mă-NĬNG-gō-sēl, p. 325)
meningomyelocele (mă-nĭng-gō-MĪ-ĕ-lō-sēl, p. 326)
myelodysplasia (mī-ă-lō-dĭs-PLĂ-zhă, p. 325)
neonatal abstinence syndrome (p. 344)
Ortolani’s sign (p. 330)
Pavlik harness (p. 331)
phototherapy (p. 339)
RhoGAM (p. 339)
shunt (p. 323)
spica cast (p. 331)
spina bifida (SPĪ-nă BĬF-ĭ-dă, p. 325)
transient tachypnea of the newborn (TTN) (p. 343)
transillumination (p. 322)
 http://evolve.elsevier.com/Leifer
http://evolve.elsevier.com/Leifer
Birth defects, abnormalities that are apparent at birth, occur in 3% to 4% of all live births. The rate is even higher if the defects that become evident later in life are counted. An abnormality of structure, function, or metabolism may result in a physical or mental disability, may shorten life, or may be fatal. Box 14-1 shows the system of classification of birth defects. Because these disorders include so many conditions, it is necessary to limit the number discussed in this chapter and to place others in relevant areas of the text (see the Index for specific conditions). Fetal alcohol syndrome and environmental influences on fetal growth are discussed in Chapter 5. Congenital heart disease is discussed in Chapter 26.
Defects present at birth often involve the skeletal system; limbs may be missing, malformed, or duplicated. Some abnormalities (e.g., congenital hip dysplasia) are more subtle, and the nurse must be alert to detect them. Inborn errors of metabolism include a number of inherited diseases that affect body chemistry. There may be an absence or a deficiency of a substance necessary for cell metabolism. The deficient substance is usually an enzyme. Almost any organ of the body may be damaged. Examples of inborn errors of metabolism include cystic fibrosis and phenylketonuria (PKU). In disorders of the blood, there is a reduced or missing blood component or an inability of a component to function adequately. Sickle cell disease, thalassemia, and hemophilia fall into this category. Chromosomal abnormalities number in the thousands. Most involve some type of mental retardation, and others are incompatible with life. The newborn with Turner’s syndrome or Klinefelter’s syndrome may have impaired physical growth and sexual development. Perinatal injuries have many causes and are seen in various forms, the most common of which is premature birth.
As the March of Dimes Birth Defect Foundation (2006) points out, “Few birth defects can be attributed to a single cause. The majority are thought to result from an interplay between environment and heredity, depending on inherited susceptibility, stage of pregnancy, and degree of environmental hazard.” Newborns with birth defects may need to remain in the neonatal unit for an extended period of time for intensive care and treatment.
Malformations Present at Birth
The following sections discuss some congenital malformations, or those defects present at birth, according to body systems.
Nervous System
Neural Tube Defects
Neural tube defects are most often caused by failure of neural tube closure at either the cranial (top) or the caudal (lower) end of the spinal cord. These defects include hydrocephalus and spina bifida.
Hydrocephalus
Pathophysiology.
Hydrocephalus (hydro, “water,” and cephalo, “head”) in the newborn is a condition characterized by an increase of cerebrospinal fluid (CSF) within the ventricles of the brain, which causes pressure changes in the brain and an increase in head size. It results from an imbalance between the production and absorption of CSF or improper formation of the ventricles. Hydrocephalus may be congenital or acquired. It is most commonly acquired by an obstruction, such as a tumor, or as a sequela of infections (encephalitis or meningitis) or perinatal hemorrhage. The symptoms depend on the site of obstruction and the age at which it develops.
Hydrocephalus is classified as noncommunicating (obstructive) or communicating. Noncommunicating hydrocephalus results from the obstruction of CSF flow from the ventricles of the brain to the subarachnoid space. Communicating hydrocephalus results when CSF is not obstructed in the ventricles but is inadequately reabsorbed in the subarachnoid space (Figure 14-1).
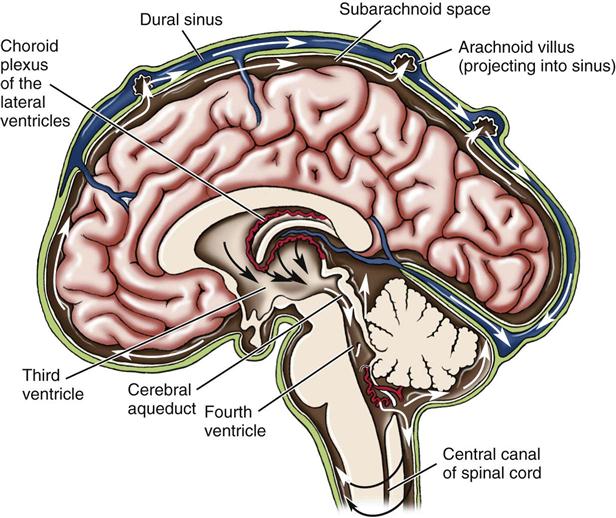
Manifestations.
The signs and symptoms of hydrocephalus depend on the time of onset and the severity of the imbalance. The classic sign is an increase in head size. If hydrocephalus occurs in utero, the enlarged head will necessitate a cesarean section delivery. At birth the head enlarges rapidly and the fontanelles bulge. The cranial sutures separate to accommodate the enlarging mass. The scalp is shiny, and the veins are dilated. In advanced cases the pupils of the eyes may appear to be looking downward and the sclera may be seen above the pupils, much like the look of a setting sun (Figure 14-2). A foreshortened occiput suggests pathology of the fourth ventricle, with the brain stem protruding through the cervical canal. This is called the Chiari malformation (Kliegman et al., 2007). When the enlarged head involves a prominent occiput, the condition usually involves an atresia of the foramen of Lushka and the foramen of Magendie and is known as the Dandy-Walker syndrome. The infant is helpless and lethargic. The body becomes thin, and the muscle tone of the extremities is often poor. The cry is shrill and high pitched. Irritability, vomiting, and anorexia are present, and convulsions may occur.
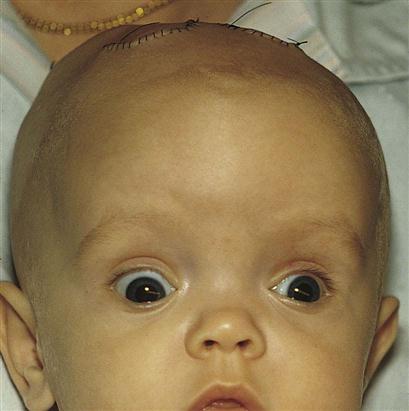
When hydrocephalus occurs in the older child, the head cannot enlarge because the cranial sutures are fused; therefore headache is the predominant symptom, with cognitive slowing, personality changes, spasticity, and other neurological signs.
Diagnosis.
Transillumination (trans, “across,” and illuminare, “to enlighten”)—the inspection of a cavity or organ by passing a light through its walls—is a simple diagnostic procedure useful in visualizing fluid. A flashlight with a sponge-rubber collar is held tightly against the infant’s head in a dark room. The examiner observes for areas of increased luminosity. A small ring of light is normal, but a large halo effect is not. The child’s head is measured daily. Echoencephalography, computed tomography (CT) scanning, and magnetic resonance imaging (MRI) are used to visualize the enlarged ventricles and to identify the area of obstruction. A ventricular tap or puncture may be performed using sterile technique to determine pressure and drain CSF. The equipment needed is the same as that for a lumbar puncture. A specimen is labeled and sent to the laboratory for analysis.
Treatment.
The use of acetazolamide and furosemide reduces the production of CSF and may provide some relief, but most often surgery is indicated (Kliegman et al., 2007). The surgeon attempts to bypass, or shunt, the point of obstruction. The CSF may thus be carried to another area of the body, where it is absorbed and finally excreted. This is accomplished by inserting special tubing, which is replaced at intervals as the child grows. The procedure, known as a ventriculoperitoneal shunt (Figure 14-3), allows the excess fluid to drain into the peritoneal cavity, where it is absorbed.
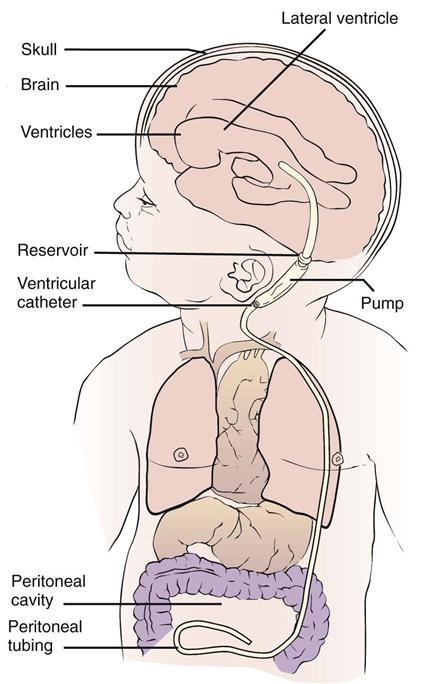
The prognosis for the child with hydrocephalus has improved with modern drugs and surgical techniques. If the brain is not seriously damaged before the operation, mental function may be preserved. Motor development is sometimes slower if the child cannot lift the head, normally because of its weight. Complications of shunts are usually mechanical (kinking or plugging of tubing) or infectious. The shunt acts as a focal spot for infection and may need to be removed if infection persists.
Preoperative Nursing Care.
The general nursing care of an infant with hydrocephalus who has not undergone surgery presents several challenges. The child may be barely able to raise the head. Mental development is delayed. Lack of appetite, a tendency to vomit easily, and poor resistance to infections complicate the management of these infants.
The position of the infant must be changed frequently to prevent hypostatic pneumonia and pressure sores. Hypostatic pneumonia occurs when the circulation of the blood in the lungs is poor and the infant remains in one position too long. It is particularly prevalent in infants who are poorly nourished or weak or who have a debilitating disease. When the nurse turns an infant who has hydrocephalus, the head must always be supported. To turn the infant in bed, the weight of the head is borne in the palm of one hand, and the head and body are rotated together to prevent a strain on the neck. When the infant is lifted from the crib, the head must be supported by the nurse’s arm and chest.
The tissues of the head, ears, and bony prominences tend to break down. A pad of lamb’s wool or sponge rubber placed under the head may help to prevent these lesions. If the skin becomes broken, it is given immediate attention to prevent infection. The infant must be kept dry, especially around the creases of the neck, where perspiration may collect.
In most cases the nurse may hold the infant for feedings. The nurse sits with the arm supported because the infant’s head is heavy. A calm, unhurried manner is necessary. The room should be as quiet as possible. After the feeding the infant is placed in a side-lying position. The infant is not disturbed once settled, because vomiting occurs easily. The nurse must organize daily care so it does not interfere with meals.
Observations to be recorded and reported include the type and amounts of food taken, any vomiting, the condition of the skin, motor abilities, restlessness, irritability, and changes in vital signs. Fontanelles are inspected for size and signs of bulging. Head circumference is measured around the occipitofrontal area and is recorded on the chart.
Symptoms of increased pressure within the head are an increase in blood pressure and a decrease in pulse rate and respirations. Signs of a cold or other infection are immediately reported to the nurse in charge and are recorded.
Postoperative Nursing Care.
In addition to routine postoperative care and observations, the nurse observes the patient for signs of increased intracranial pressure (ICP) and of infection at the operative site or along the shunt line. As with any postoperative care, pain control management is essential.
Bacterial infection is a life-threatening complication that sometimes necessitates shunt removal. Signs of infection include an increase in vital signs, poor feeding, vomiting, pupil dilation, decreased levels of consciousness, and seizures. The operative area is observed for signs of inflammation. An internal flushing device may be used to ensure patency of the shunt tube when increased ICP is suspected. The surgeon may order the pump to be routinely depressed a certain number of times each day to facilitate drainage. This is accomplished by compressing the antechamber or reservoir that is under the skin behind the ear (see Figure 14-3).
Positioning of the infant depends on several factors and may vary with the infant’s progress. If the fontanelles are sunken, the infant is kept flat because too rapid a reduction of fluid may lead to seizures or cortical bleeding. If the fontanelles are bulging, the infant is usually placed in the semi-Fowler’s position to promote drainage of the ventricles through the shunt. The infant is always positioned in a way that prevents pressure on the operative site. The surgeon leaves orders for the patient’s position and activity. Assessment of skin remains a priority. Head and chest measurements are recorded. In patients with peritoneal shunts, the abdomen is measured or observed to detect malabsorption of fluid.
The infant should be observed for signs of increased ICP. The development of a high-pitched cry, unequal pupil size or response to light, bulging fontanelles, irritability or lethargy, poor feeding, or abnormal vital signs should be reported and recorded. (Evidence of increased ICP in the older child may be manifested by a change in personality, a change in level of consciousness, and complaints of headache that is unrelieved by over-the-counter medications.) The need for pain control should be assessed and medications given as needed. Intake and output are carefully recorded, and the infant is observed closely for signs of fluid overload. The infant is usually fed after active bowel sounds are heard. The surgical suture lines should be kept clean and dry and the infant’s diaper kept well below the abdominal suture line to prevent contamination.
Parent education, support, and guidance are essential. Parents are taught signs that indicate shunt malfunction, how and when to “pump” the shunt by pressing against the valve behind the ear, and the need for multidisciplinary follow-up care. Signs of tube malfunction in the older child involve signs of increasing ICP such as headache, lethargy, and changes in level of consciousness. Community resources, such as the National Hydrocephalus Foundation and information concerning special car seats for children with special needs, should be made known to the parents. There is approximately an 80% survival rate for infants treated early, and approximately one third of the cases result in normal physical and neurological functioning. Other survivors may have varying degrees of developmental disabilities.
Spina Bifida
Spina bifida, also known as myelodysplasia, refers to a group of central nervous system disorders characterized by malformation of the spinal cord.
Pathophysiology.
Spina bifida (divided spine) is a congenital embryonic neural tube defect in which there is an imperfect closure of the spinal vertebrae. There are two forms: occulta (hidden) and cystica (sac or cyst) (Figure 14-4).
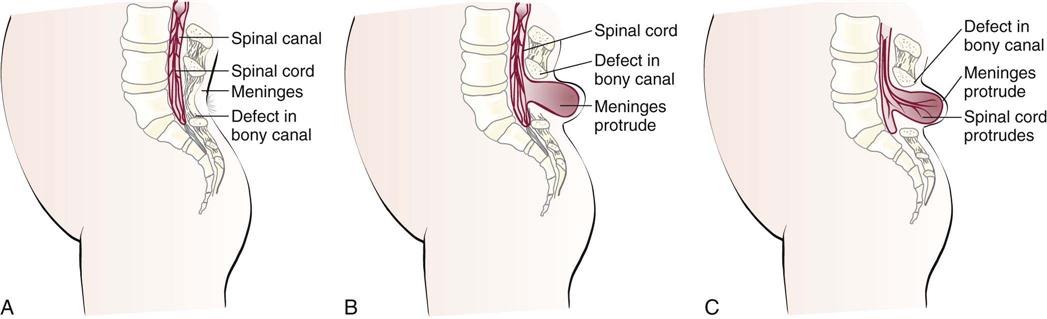
Spina bifida occulta is a relatively minor variation of the disorder in which the opening is small and there is no associated protrusion of structures. It often goes undetected and occurs most commonly at the L5 and S1 levels. There may be a tuft of hair (Figure 14-5), dimple, lipoma, or discoloration at the site. In general, treatment is not necessary unless neuromuscular symptoms appear. These symptoms consist of progressive disturbances of gait, such as footdrop, or disturbances of bowel and bladder sphincter function.
Spina bifida cystica consists of the development of a cystic mass in the midline of the opening in the spine. Meningocele and meningomyelocele are two types of spina bifida cystica. A meningocele (meningo, “membrane,” and cele, “tumor”) contains portions of the membranes and CSF. The size varies from that of a walnut to that of a newborn’s head.
More serious is a protrusion of the membranes and spinal cord through this opening, or a meningomyelocele. Although it resembles a meningocele, there may be associated paralysis of the legs and poor control of bowel and bladder functions. Hydrocephalus is a common complication. Prenatal detection is possible through ultrasonography and testing for increased alpha-fetoprotein (AFP) in the amniotic fluid of the mother (see Chapter 5).
Prevention.
The specific cause of meningomyelocele is unknown. The use of drugs during early pregnancy and poor nutrition may contribute to the development of a neural tube defect. The American Academy of Pediatrics (AAP) recommends that all women of childbearing age take a daily multivitamin that contains 0.4 mg of folic acid and continue the intake of folic acid until the twelfth week of pregnancy, when basic neural tube development is completed. Studies have shown that the intake of folic acid before conception dramatically decreases the occurrence of neural tube defects such as spina bifida.
 Nursing Tip
Nursing Tip
The intake of a daily multivitamin containing 0.4 mg of folic acid before conception can reduce the risk of neural tube defects such as spina bifida.
Treatment.
The treatment for spina bifida is surgical closure. The prognosis for patients with these conditions depends on the extent of involvement. When a patient with meningocele has no weakness of the legs or sphincter involvement, surgical correction is performed with excellent results. Surgery is also indicated for a patient with meningomyelocele for cosmetic purposes and to help prevent infection. A multidisciplinary approach is necessary because, depending on the extent of the defect, the child may have difficulties associated with hydrocephalus, orthopedic problems, and problems relating to urinary and bowel function.
Habilitation is necessary after the operation because the legs remain paralyzed and the patient is incontinent of urine and feces. Habilitation, rather than rehabilitation, is the term used to describe this treatment because the patient is disabled from birth and therefore is learning, not relearning. The aim of habilitation is to minimize the child’s disability and put to constructive use the unaffected parts of the body. Every effort is made to help the child develop a healthy personality so that he or she may experience a happy and productive life.
Eventually the child can be taught to use a wheelchair and possibly to walk with braces, crutches, or other walking devices. The implantation of an artificial urinary sphincter in early childhood can help some children to become continent and prevent the complications associated with constant urinary dribble. Medications such as oxybutynin chloride (Ditropan) are available to increase bladder storage. Children can also be “bowel trained” with the use of suppositories that promote timed bowel movements, helping the child avoid the social rejection that can be caused by bowel incontinence.
Nursing Care.
The main objectives of nursing care include prevention of infection of or injury to the sac, correct positioning to prevent pressure on the sac and development of contractures, good skin care (particularly if the infant is incontinent of urine and feces), adequate nutrition, accurate observations and charting, education of the parents, continued medical supervision, and habilitation.
Immediate care of the sac is essentially the same regardless of whether the cord is involved. On delivery the newborn is placed in an incubator. Moist, sterile dressings of saline or an antibiotic solution may be ordered to prevent drying of the sac. Some method of protecting the mass is necessary if surgery is to be delayed. Protection from injury and maintenance of a sterile environment for the open lesion are essential.
Along with routine observations made for every newborn, other pertinent nursing observations must be made and recorded:
• The size and area of the sac are checked for any tears or leakage.
• The head circumference is measured to determine the possibility of associated hydrocephalus.
• Fontanelles are observed to provide baseline data.
• The lack of anal sphincter control and dribbling of urine are significant in the differential diagnosis (Figure 14-6). In general, the higher the defect on the spine, the greater the neurological deficit.
Positioning of the infant is of importance. The goal is to prevent pressure on the sac and prevent postural deformities. When positioning infants with multiple deformities, the nurse must try to guard against aggravating existing problems. The infant is usually placed prone with a pad between the legs to maintain abduction and to counteract hip subluxation. A small roll is placed under the ankles to maintain foot position. Some infants may be supported in a side-lying posture to provide periods of relief. The disadvantage of this position is that it reduces movement of the arms and flexes the hips. The physical therapy staff may provide a helpful consultation. Surgery is generally done early.
Postoperative nursing care involves neurological assessment and prevention of infection. The status of the fontanelles and any signs of increased ICP, such as irritability or vomiting, are significant. Sometimes a shunt is performed shortly after closure of the spine, if hydrocephalus is present. Complications that can be life threatening include meningitis, pneumonia, and urinary tract infection.
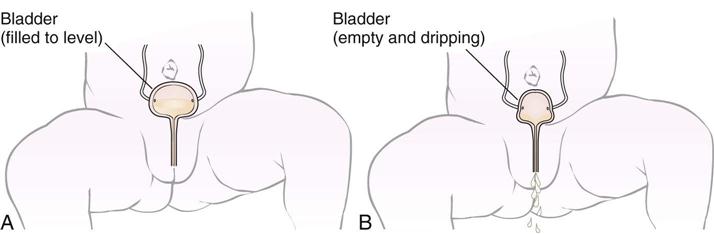
Urological monitoring is essential, because many of these infants have urinary incontinence (see Figure 14-6). Medication to prevent urinary tract infections is routinely given. The Credé method of bladder emptying (applying pressure above the symphysis pubis) may be used for infants. Older children may be taught intermittent, clean self-catheterization. This technique can be performed by parents and learned by children.
Skin care is a challenge. Constant dribbling of feces and urine irritates the perineal area and can infect the sac or the incision. Meticulous cleanliness is necessary. The bedding must be dry and free of wrinkles. Frequent cleansing, application of a prescribed ointment or lotion, and light massage help to maintain skin integrity. If range of motion exercises are ordered, they are performed gently.
Feeding is facilitated by early closure of the defect. In delayed cases, gavage may be used. These patients need cuddling and sensory stimulation. An infant who cannot be held can be soothed by touch. The nurse talks to the infant and, when possible, provides face-to-face (en face) communication. Mobiles are placed appropriately. Periodically moving the incubator or crib provides diversity of view. Soft music is also soothing.
Many infants with spina bifida develop a latex allergy. A latex-free environment should be adopted whenever possible. Parents should be informed that latex products such as balloons, “koosh” balls, tennis balls, and adhesive strips can cause allergic reactions that may include rashes and wheezing. The parents should be informed about food sensitivities that are common to children with latex allergies. Foods to avoid include bananas, avocados, and kiwi. Other commonly used items to avoid include latex-based pacifiers, feeding nipples, and water toys. The child should wear a medical identification tag indicating the latex allergy. In some cases, antihistamines and steroids may be prescribed before and after surgery. Nurses should wear nitrile gloves instead of latex gloves while caring for these patients.
Special consideration must be given to the establishment of parent-infant relationships. This problem is complicated if the infant is transferred to a large medical center. Understanding and support are given to the parents, who may be overwhelmed. It is not unusual for them to be repulsed by the cyst. Most experience a sense of loss for what was to have been their “perfect baby.” Steps of the grieving process may be recognized by the astute nurse. Information and education about this disorder can be obtained from the Spina Bifida Association of America.
Gastrointestinal System
Cleft Lip
Pathophysiology.
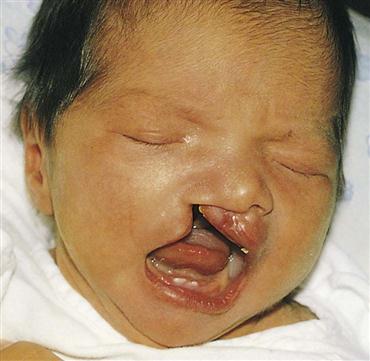
A cleft lip is characterized by a fissure or opening in the upper lip (Figure 14-7). It is a result of the failure of the maxillary and median nasal processes to unite during embryonic development, usually between the seventh and eighth weeks of gestation. In many cases it seems to be caused by hereditary predisposition, but occasionally it can be caused by environmental influences during the stage of oral development. This disorder appears more frequently in boys than in girls and may occur on one or both sides of the lip. The extent of the defect may vary from slight to severe. Sometimes it is accompanied by a cleft palate—a fissure in the midline of the roof of the mouth. Cleft lip and cleft palate are common congenital anomalies and occur in about 1 in 750 births. Transculturally, they occur more often in Asian Americans and Native Americans and less commonly in African Americans (Kliegman et al., 2007).
Treatment and Nursing Care.
The initial treatment for cleft lip is a surgical repair known as cheiloplasty. The cleft lip is repaired by age 3 months when weight gain is established and the infant is free of infection. Surgery not only improves the infant’s sucking ability but also greatly improves appearance. Indirectly, this influences bonding and the amount of affection the infant receives because some parents refrain from cuddling an infant who is obviously disfigured.
A complete physical examination is done and routine blood tests are ordered before surgery. Photographs may also be taken. Any signs of oral, respiratory, or systemic infection are reported to the registered nurse. The physician may order elbow restraints to prevent the infant from scratching the lip and to acquaint the infant with them because they are necessary postoperatively. A syringe with a rubber tip, a long nipple with a large hole attached to a squeeze bottle, or a medicine dropper can be used to feed the infant before and after surgery, because sucking motions must be avoided to keep from applying tension on the suture line.
Postoperative Nursing Care.
Postoperative nursing goals for the infant undergoing a cheiloplasty include the following:
Feeding.
The infant receives feedings by dropper until the wound is completely healed (1 to 2 weeks). The infant is usually fed as soon as clear liquids are tolerated postoperatively. Care should be taken to avoid touching the suture line when inserting the medicine dropper. Sucking is prevented as much as possible until the suture line is healed. Placing a small amount of formula into the infant’s mouth and allowing time for swallowing will prevent aspiration. Offering small amounts of sterile water will cleanse the mouth after feeding. Formula or drainage is gently cleaned from the suture line with saline solution, and an ointment may be applied to the skin as prescribed. Holding the infant during feedings, burping frequently, and placing the infant in an infant seat after feeding or on the right side propped with a rolled blanket will aid in a positive outcome for this infant. The mother who has fed her infant preoperatively and has been allowed to assist with feedings during hospitalization will feel more confident after discharge. The immediate improvement as a result of surgery is encouraging to the parents, particularly if the child must have further surgery for cleft palate repair.
Cleft Palate
Pathophysiology.
A cleft palate is a failure of the hard palate to fuse at the midline during the seventh to twelfth weeks of gestation. This separation forms a passageway between the nasopharynx and the nose, which not only complicates feeding but also easily leads to infections of the respiratory tract and middle ear that can result in hearing loss. It is generally responsible for speech difficulties that occur in later life. The cleft may not be readily apparent at birth, and for this reason careful examination of the oral cavity and upper palate at birth is essential. Feeding is a problem because the cleft prevents negative pressure from being formed within the mouth, which is necessary for successful sucking.
Treatment.
The goals of therapy are union of the cleft, improved feeding, improved speech, improved dental development, and the nurturing of a positive self-image. Some surgeons prefer to operate between 1 year and 18 months of age if at all possible so that speech patterns are minimally affected. If surgery has been deferred, a dental speech appliance may be used to facilitate communication. This appliance must be changed periodically as the child grows.
Treatment of the child with a cleft lip and palate requires multidisciplinary teamwork with a surgeon, pediatrician, pediatric dentist, orthodontist, nurse, psychologist, speech therapist, and social worker. The public health nurse should be responsible for coordinating parental counseling and referral as needed. The emotional problems that sometimes occur with this condition require more extensive attention than does the repair itself. A child born with a facial deformity encounters many problems. Feedings are difficult and are not relaxed in the initial period. As the child grows, irregular tooth eruptions, drooling, delayed speech, and the need for intermittent hospitalization and frequent clinic appointments can be frustrating. High-resolution ultrasound can detect cleft palate by 13 weeks of gestation, and therefore its correction (without scarring) by fetal surgery appears promising for the near future.
 Safety Alert!
Safety Alert!
Suctioning the mouth should be avoided in infants who have a cleft palate repair.
Psychosocial Adjustment of the Family.
A mother’s first reaction to a disfigured newborn is one of shock, hurt, disappointment, and guilt. Some parents regard the deformity as a result of their inadequacies. They may desire to hide the child from relatives and friends. The developing child senses the parents’ feelings and acquires either a positive or a negative self-image. The patient and family need understanding, a concrete basis for hope, and practical advice. Family stress often occurs because of the multiple surgeries that may be required throughout childhood.
Follow-up Care and Home Care.
In large cities, special cleft palate clinics are available in which several specialists can work together in convenient consultation. The parents are instructed about the resources available in the state in which they live. The American Cleft Palate–Craniofacial Association, the Cleft Palate Foundation, the March of Dimes Birth Defect Foundation, and state programs for children with special needs are examples of community referrals that should be offered to parents.
Postoperative Treatment and Nursing Care
Nutrition.
Fluids are taken by a cup, although a gravity feeder may be desirable in some cases. The method varies with the plastic surgeon. The diet is progressive, at first consisting of clear fluids and then full fluids. By the time of discharge, a soft diet can generally be taken. Hot foods and liquids are avoided to prevent injury to the operative site. The patient must not suck on a straw. When feeding with a spoon, the nurse should place the spoon into the side of the mouth. The spoon must not touch the roof of the mouth. The nurse teaches parents to keep objects such as the child’s thumb, tongue blades, toast, cookies, forks, and pacifiers out of the mouth. Elbow restraints are used to prevent the child from placing his or her fingers or objects in the mouth. The diet is advanced only on consultation with the physician.
Oral hygiene.
The mouth is kept clean at all times. Feedings are followed by a little water. The physician may prescribe a mild antiseptic mouthwash.
Speech.
It is helpful to speak slowly and distinctly to the child. The child is encouraged to pronounce words correctly. Children who have undergone extensive repairs or have associated deafness need the help of a speech therapist. The speech therapist evaluates the child and assists the parents in specific activities that facilitate speech development.
Diversion.
Crying is to be prevented as much as possible. Play should be quiet, particularly in the immediate postoperative period. The nurse reads, draws, or colors with the child.
Complications.
Ear infections and dental decay may accompany cleft palate. Parents are instructed to take the child to the health care provider at the first sign of earache. Regular visits to the dentist are scheduled. Throughout the long-term care, a stable goal in the care of this infant is to promote optimal growth and development and to establish positive self-esteem.
Musculoskeletal System
Clubfoot
Pathophysiology.
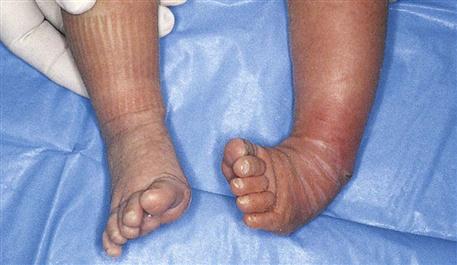
Clubfoot, one of the most common deformities of the skeletal system, is a congenital anomaly characterized by a foot that has been twisted inward or outward. The incidence is about 1 in 1000 live births. Many mild forms are caused by improper position in the uterus, and these clear up with manipulative exercises. In contrast, true clubfoot does not respond to simple exercise. Several types are recognized. Talipes (talus, “heel,” and pes, “foot”) equinovarus (equinus, “extension,” and varus, “bent inward”) is seen in 95% of patients. The feet are turned inward, and the child walks on the toes and the outer borders of the feet. It generally involves both feet (Figure 14-8).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


