CHAPTER 12 1. Define the concepts of (a) pharmacology, (b) pharmacodynamics, and (c) pharmacokinetics. 2. Describe the developmental changes that affect medication absorption, distribution, metabolism, and elimination. 3. Identify specific considerations when one is administering medications of the following types to a neonate: (a) antimicrobial agents, (b) cardiovascular agents, (c) central nervous system agents, (d) diuretics, and (e) immunizations. 4. Describe nursing responsibilities and interventions when administering medication to the neonate. The study and clinical application of neonatal pharmacology can facilitate safe medication administration in the neonate. The application of pharmacologic principles involves evaluating existing knowledge related to the pharmacodynamic and pharmacokinetic responses of the neonate to specific medications. Unfortunately, this body of knowledge is extremely limited in the neonatal population owing to a lack of controlled clinical trials with these patients. Confounding factors relative to gestational and chronologic age, weight, fluid status, and the health–illness state of individual organ systems make dosing medications in this population a challenging and perpetual learning process. The decision to administer a medication should be evaluated for the desired response and the potential for an undesirable reaction. The nurse is in the ideal position to observe and evaluate both response and reaction and to intervene if necessary. This chapter provides pharmacologic information specific to the neonate. Information on medication dosages and implications for medication administration is provided in individual clinical chapters. Additional current reference materials should also be available in the neonatal intensive care unit (NICU). A. Pharmacology: the science of the properties of medications and their effects in the body. 2. Medication: any substance or mixture of substances intended to be used for the cure, mitigation, or prevention of disease in human beings or animals. 3. Pharmacodynamics: the relationship between medication concentrations at the site of action and the pharmacologic response (intensity and time course of therapeutic and adverse effects). This is what the drug does to the body. 4. Pharmacokinetics: the fate of a medication in the body from the time it enters until it and all of its metabolites are removed. This includes medication absorption, distribution, metabolism, and excretion. It is also the specialized study of the mathematical relationship between a medication dosage regimen and the resulting serum concentration. This is what the body does to the drug. 5. Bioavailability: the portion of the administered dose that reaches the site of action in the body. This is usually the amount entering the circulation and may be reduced when medications are given by mouth versus when given intravenously. 6. Therapeutic range: a range of medication concentrations within which the probability of the desired clinical response is relatively high and the probability of unacceptable toxicity or subtherapeutic response is relatively low. 7. Therapeutic drug monitoring (TDM): determinations of plasma medication concentrations to optimize medication therapy. TDM is valuable when: a. A good correlation exists between the pharmacologic response and plasma concentration. b. Wide intersubject variation in drug plasma levels results from a given dose. c. The medication has a narrow therapeutic range. d. The medication’s desired pharmacologic effects cannot be readily assessed by other simple means. 8. Steady state: a term used to refer to a situation in which the amount of medication administered is equal to the amount of medication eliminated. When steady state is reached in a patient, the blood concentrations remain “steady.” Therefore, at steady state, all peak drug levels and all trough drug levels should be the same. 9. Half-life: the time necessary for a measured medication concentration to fall to half its original value. A medication’s duration of action is often related to its half-life, and the half-life may also indicate when another dose should be given. It takes approximately five half-lives to reach steady state. 10. Types of medication levels: b. Trough level: a drug level that is drawn just prior to the dose. c. Random level: a drug level that is drawn at any time after a dose is given. These levels are often used to follow drug levels in patients with changing renal function or changing volume status. 2. The individual characteristics of the receptor are responsible for the selective nature of medication response. 3. Receptor theory of medication action allows an explanation of medication antagonists. The antagonist medication may alter the characteristics of the receptor molecule in a way that limits or inhibits the response to the original medication (e.g., naloxone and morphine) or stimulus (e.g., a β-blocker such as propranolol). 4. Some medications do not appear to act through receptors. Their action is related to a direct response in the recipient. B. General mechanisms of medication action. 1. Based on the nature of the receptor–medication complex. 2. Types of receptor–medication complexes. a. Receptor–medication complexes that regulate gene expression. (2) These medications generally do not have a rapid effect after initial administration (e.g., epoetin alfa). b. Receptor–medication complexes that change cell membrane permeability. (2) These medications may have a relatively short lag time between administration and response (e.g., penicillin). c. Receptor–medication complexes that increase the intracellular concentration of a second messenger molecule. (1) These medications increase production and activity of enzyme systems within the cell. (2) These medications may stimulate a rapid response in changing cell characteristics (e.g., dopamine). C. Relationship between medication dose and clinical response. b. Extreme sensitivity: a patient whose response to a medication is more intense than is expected (e.g., severe hypotension from an antihypertensive agent). c. Unpredictable adverse reaction: a patient whose medication reaction is substantially different from what would have been predicted and may differ from the usual response in most patients (e.g., an anaphylactic reaction). d. Tolerance: a diminished response to a given medication dose that is related to long-term administration of a medication (e.g., fentanyl doses must be increased as the length of therapy increases to achieve the same effects). e. Tachyphylaxis: a rapidly diminished medication response without a medication dosage change. This may be caused by any of a number of factors, including a limited number of receptor sites or limited numbers of transmitter chemicals (e.g., response to albuterol may diminish if given frequently). 2. Factors that may affect individual medication response are as follows: b. Variation in amounts of antagonistic substances: an unusually large or limited amount of antagonistic substances that alter receptor molecule response. c. Alterations in numbers or function of receptor molecules: an increased or diminished number of receptor molecules changes the number of potential medication–receptor complexes. d. Changes in concentration of molecules other than receptor molecules: if medication response is ultimately dependent on an effect on molecules other than those of the medication–receptor complex, medication response may be limited by the amount of the third molecule type (e.g., prodrugs such as fosphenytoin). D. Desired versus undesired effects of medications. b. Subtherapeutic effect: those effects that are less than the desired outcome of the medication administration (e.g., continued apnea episodes with low theophylline levels). c. Side effects: those medication effects that result from medication administration and that are in addition to the desired effects. All medications have some side effects, varying from minor and clinically insignificant to major side effects that are sufficiently adverse to require discontinuation of the medication therapy (e.g., tachycardia with theophylline treatment). d. Toxic effects: medication response that results from a medication overdose or unexpected high serum medication concentrations (e.g., seizures from a high theophylline level). 2. It is the responsibility of the health care provider to weigh the benefits of the therapeutic effect against the risk of undesirable side effects and toxic effects or subtherapeutic responses and make adjustments accordingly. A. Principles of medication absorption (Fig. 12-1). 1. General principles of medication absorption. a. The movement of a medication from the site of administration to the bloodstream. b. Regardless of the route of administration, most medications must cross cell membranes to reach their site of action. (2) Medications can enter the cell through other mechanisms also, such as active transport or facilitated diffusion. c. The absorption of a medication is dependent, in large part, on the route of administration. The common sites of administration are as follows: (a) Gastric emptying and intestinal transit times: prolonged and irregular; approaches adult values at 6 to 8 months of age. Gastric and intestinal motility are reduced with prematurity, asphyxia, gastroesophageal reflux, and respiratory distress syndrome. Administration of hypocaloric feeds, human milk, or prokinetic agents (e.g., metoclopramide, erythromycin) can increase motility. Prolonged transit times and enterohepatic recirculation may also increase the bioavailability and pharmacologic effect of some substances (Chemtob, 2011; van den Anker et al., 2011). However, the bioavailability of the medication may decrease because of increased first-pass loss, increased GI destruction of medication or shortened transit times from diarrhea or emesis. (b) GI tract acidity: the presence of marked changes in pH from the stomach through the distal portion of the GI tract may affect absorption. The pH of the GI tract is nearly neutral or slightly alkaline at birth owing to the presence of amniotic fluid in the stomach. Gastric acid increases secretion rapidly such that it doubles by 2 months, even in the extremely low birth weight neonate (Kearns et al., 2003; van den Anker et al., 2011). The net effect of GI acidity on the absorption of medications is dependent on the pH characteristics of the medication and preparation. Medications that normally may not be absorbed well in the stomach may be absorbed at a higher rate in the neonate because of decreased stomach acidity. The absorption of acidic drugs will be reduced (e.g., phenobarbital, phenytoin) and the absorption of basic drugs will be enhanced (e.g., penicillin, erythromycin) as compared with adults (Funk et al., 2012). (c) GI enzyme activity: neonates are deficient in pancreatic enzymes at birth. This deficiency may inhibit absorption of some medications that require pancreatic enzymes for efficient absorption. One compensating mechanism that neonates have is an increased β-glucuronidase secretion. This is an enzyme produced by organisms normally present in the small intestine that are capable of metabolizing some medications. It is present in neonates at up to seven times the adult amounts (Kearns et al., 2003; van den Anker et al., 2011). (d) Bacterial flora: composition and rate of colonization of the GI tract by the normal bacterial flora may affect both GI tract motility and the metabolism of some medications. Colonization is dependent on oral intake, antibiotic administration, or disease states such as necrotizing enterocolitis or infectious gastroenteritis. Normal colonization in vaginally born term neonates occurs by 4 to 6 days of age. Intestinal flora are required for the production of vitamin K (Chemtob, 2011). (e) GI tract perfusion: in very ill neonates, hypoperfusion of the gut may decrease medication absorption. (f) Underlying disease states: underlying disease states such as diarrhea, emesis, or the presence of nasogastric suction may reduce the time available for medication absorption. Diseases of genetic (e.g., cystic fibrosis) or circulatory (e.g., necrotizing enterocolitis) origin will alter pancreatic enzymes or intestinal mucosa, decreasing GI absorption. (2) Rectal: may be a very rapid and efficient means of medication absorption. (b) Serum levels of some medications may be as high as levels obtained through the IV route of administration (Kearns et al., 2003). (c) Routine administration by rectal route is discouraged. Relative volume and fragility of the neonatal rectum must be considered. (d) The retention time is the rate-limiting step. (3) Inhalation: useful for gaseous or easily vaporized medications. (b) Medications administered by this route have a particular advantage when the site of desired action is the tracheobronchial tree (e.g., albuterol). (c) There is a potential for fewer side effects when medications are administered this way, because much of the medication may not be systemically absorbed (e.g., inhaled corticosteroids). (d) Most frequently used in neonatal intensive care for the administration of various surfactant preparations. (e) Certain medications (e.g., epinephrine and lidocaine) can be given through the endotracheal tube in emergency situations when IV access is not readily available. (f) This route may be less effective in a neonate with pulmonary hypertension and poor or abnormally distributed pulmonary blood flow. (4) Topical/percutaneous: utility is limited to medications whose absorptive characteristics allow permeation through the skin or mucous membranes. Percutaneous absorption has particular advantages as well as risks in the neonate. (a) The rate of absorption is inversely related to skin thickness and directly related to skin hydration. With increasing gestational age, skin thickness increases and water content decreases, thus reducing the amount of absorption from this route. Maturation of the epidermis occurs between 23 and 33 weeks of gestation. The stratum corneum forms after 23 to 24 weeks of gestation. Formation of this layer of the skin greatly decreases the permeability of the skin to water and infection (Hoath, 2011). This allows much more efficient percutaneous absorption of medications in neonates of lower birth weight and younger gestational age. However, this poses a particular hazard in care, because substances that may be safely applied to the skin of a more mature patient may be absorbed in dangerous amounts in the immature neonate (van den Anker et al., 2011). (b) The ratio of body surface area to body weight is higher in neonates than adults, providing a relatively larger absorptive surface in comparison with body mass. (c) The absorptive response time is variable and may be limited to the local area of application; however, some topically applied medications (e.g., nitroglycerin) can have a systemic effect. As a result, products that contain alcohol and hexachlorophene skin washes should be avoided. Any treatment with topical steroids should be limited to less than 2 weeks to prevent adrenal suppression. On the other hand, antibiotic ointments will be more readily absorbed to treat infections, povidone–iodine (Betadine) absorption can prevent iodine deficiency, and topical safflower oil may prevent essential fatty acid deficiency. (d) Occlusive dressings will increase the extent of absorption. (5) Intramuscular/subcutaneous: administration into muscle or subcutaneous tissue. (b) These routes are limited to medications that do not cause tissue damage at the administration site and are soluble at physiologic pH. (c) These routes will achieve slower responses as compared with the IV route because of the lag time between administration and achievement of blood concentration. (d) Response time is dependent on blood flow to muscle and may be greatly delayed in hypoperfused tissue. Poor peripheral blood flow and low blood pressure are common in neonates. These problems become less common with increasing gestational age but can occur in the presence of many neonatal disease states. Poor cardiac output frequently occurs with many illness states in the neonatal period. Subsequent increases in the peripheral perfusion after resolution of the primary illness states may put the neonate at risk of having an increase in the rate and amount of medication absorption. (e) Diminished muscle activity in the ill neonate decreases muscle perfusion and consequently may limit absorption of medications administered by this route. (6) Intravenous: direct administration into the bloodstream. (a) Bypasses all absorptive barriers. (b) Most effective and reliable method of medication administration because the medication is delivered directly to the circulating plasma volume. (c) Significant medication serum concentrations are reached rapidly, allowing for immediate medication response. This includes both desired and undesired or toxic reactions. (d) Adequate and equal distribution to all organs or compartments is not guaranteed. Characteristics of some biological membranes may limit medication distribution to body compartments (e.g., blood–brain barrier). (e) Rapid achievement of potentially dangerous serum medication concentrations may require administration of the IV medication over a prolonged period rather than by IV push (e.g., vancomycin, amphotericin, and aminoglycosides). B. Principles of medication distribution. 2. Body compartments. a. Total body water: as age increases, total body water, as a percentage of total body mass, decreases. Total body water in adults comprises approximately 55% of total body weight; in term infants it is 75%, and in preterm infants it is 85% (Funk et al., 2012). (2) Because of increased total body water, a less mature neonate may require a larger per-kilogram dose to achieve the same peak medication concentration and effect as an older patient (e.g., aminoglycosides). (3) In the first several days after birth, total body water decreases, causing the volume of intracellular fluid to be increased relative to the amount of extracellular water (van den Anker et al., 2011). This leads to rapid changes in the volume of distribution for water-soluble medications, in relation to normal physiology and illness states. (4) Body water loss is divided into two main categories: sensible and insensible. Sensible losses can be measured and quantified. In term neonates, insensible losses occur in a strong relationship to metabolism. In extremely premature neonates, insensible losses occur primarily through evaporative loss, independent of metabolic rate. Insensible losses are difficult to quantify (Chemtob, 2011). (5) Medications frequently administered to the neonate (e.g., diuretics, indomethacin) may have a major impact on body water volume and, as a side effect, may alter the volume of distribution (Chemtob, 2011). (6) Neonates with disease states that alter water excretion (e.g., primary renal disease, syndrome of inappropriate antidiuretic hormone [SIADH], congestive heart failure, capillary leak syndromes) may have expansion of body water as a result of this dysfunction. (7) The preceding alterations make dosing medications that are primarily distributed in body water (e.g., aminoglycosides) difficult. (8) Frequent monitoring of medication levels may be required as the mentioned changes in body compartment volumes occur. b. Fat: wide variability of neonatal values, based on gestational age and intrauterine and postnatal growth patterns. This percentage increases from 1% of body weight at 28 weeks of gestation to approximately 15% in a term neonate (Stewart and Hampton, 1987). (1) Medications with more lipid solubility have affinity for this tissue. (2) Reduced amounts of the percentage of body fat may make the volume of distribution smaller for medications distributed primarily in fatty tissue. Hence the plasma levels of these medications will be higher because less drug will be bound in the fat tissue, resulting in a greater potential for side effects and toxicity (e.g., morphine, lorazepam). c. Blood components: potential sites for medication binding. (1) Erythrocytes: neonates have 2.5 times more binding sites for digoxin as compared with the erythrocytes in adults. This results in the need for much higher doses per total body weight in neonates than adults (Stewart and Hampton, 1987). (2) Plasma protein concentrations: medications may form a complex with large circulating molecules (usually proteins). (b) The primary binding protein for acidic medications (e.g., phenytoin, indomethacin, and furosemide) in the serum is albumin (Chemtob, 2011). (c) Serum albumin levels may be markedly decreased in the ill, extremely low birth weight neonate. (d) The primary binding proteins for basic molecules (e.g., lidocaine, propranolol) are lipoproteins, glycoproteins, and β-globulins (Chemtob, 2011). (e) Unconjugated bilirubin: many neonates have increased plasma unconjugated bilirubin levels. Fetal albumin has an increased affinity for bilirubin and a decreased affinity for medications. This causes the unconjugated bilirubin to displace some medications from the albumin-binding sites, making more free medication available for action. In contrast, some medications (e.g., ceftriaxone, sulfonamides) may displace unconjugated bilirubin from albumin-binding sites. This may lead to the deposition of unconjugated bilirubin in the neonatal brain, causing kernicterus (Kearns et al., 2003). These medications should be avoided if possible in patients younger than 2 months of age or in patients with high unconjugated bilirubin levels. (f) Free fatty acids: increased serum free fatty acid concentrations have been shown to displace some medications from plasma albumin-binding sites (Chemtob, 2011). (g) Blood pH: acidosis is a common finding associated with many neonatal disorders. Changes in blood pH have been shown to change albumin-binding characteristics. This may cause medication displacement from albumin. Changes in blood pH may also cause medications to displace unconjugated bilirubin (see above). d. Blood–brain barrier: incomplete in the neonate, causing a greater permeability to lipophilic medications (e.g., phenytoin, benzodiazepines). This incomplete barrier may be beneficial when treating neonatal meningitis because the antibiotics penetrate the cerebrospinal fluid more readily (Stewart and Hampton, 1987). 3. Medication movement: an important part of medication distribution involves medication movement from the site of administration to sites throughout the body. It is dependent on the blood flow and medication solubility. (1) Hypotension: may affect peripheral medication absorption and/or distribution. (2) Distributive shock: caused by inappropriate vasodilation; seen with sepsis. Medication distribution to specific organs may be limited by local hypoperfusion. (3) Pulmonary hypertension: may impede medication delivery to the pulmonary vascular bed. (4) Patent ductus arteriosus: blood flow may be distributed preferentially to either the pulmonary or systemic circulation, depending on the pressure differential. (5) Congestive heart failure: may affect peripheral medication absorption or distribution. b. Medication solubility: in biological tissues, the relative ability of the medication to dissolve in biological fluids. (2) The relative medication solubility may make some medication use inappropriate. A medication with low lipid solubility may not reach therapeutic concentrations in an organ that is primarily fat tissue (e.g., aminoglycosides do not readily cross the blood–brain barrier). C. Principles of medication metabolism. (1) Metabolic activity is divided into two main types. (a) Phase I (nonsynthetic) metabolism of medications: primarily oxidation, reduction, hydrolysis, or demethylation reactions, which generally occur in the smooth endoplasmic reticulum of the hepatocyte. The function of these enzymes in the full- and preterm neonate is approximately 50% to 70% of adult values. Maturation occurs as a function of chronologic rather than postconceptional age, with a wide range of variability. Maturation of nonsynthetic enzyme systems during the first several days of life requires careful monitoring of serum levels of some classes of medications (e.g., anticonvulsants). For instance, at term gestation, neonates have approximately 30% of adult ability to metabolize phenytoin, hence the half-life is significantly prolonged. Within several weeks of medication exposure, metabolic enzyme activity for phenytoin surpasses adult activity (Funk et al., 2012; van den Anker et al., 2011). (b) Phase II (synthetic) metabolism of medications: primarily involves the acetylation, methylation, or conjugation of the medication with another substance. The function of these enzymes is also immature, and they may not reach adult levels in concentration and function until well after the neonatal period. (2) Hepatic uptake of the medication is dependent on the concentration of the medication in the liver (dependent on hepatic blood flow) and the hepatocyte concentration of ligandin (Y-protein). This protein is responsible for substrate uptake by hepatic cells. (3) In the first-pass effect, hepatic biotransformation may markedly alter medication availability by directly metabolizing medications absorbed from the GI tract, before those medications reach other organs. Slow GI motility may increase the potential for first-pass effect. Prolonged GI transit times may increase potential for hepatic metabolism and eventual excretion of orally administered medications. (4) Certain medications (e.g., phenobarbital) are thought to induce enzyme maturity in the fetus and neonate, resulting in increased rates of medication elimination. (5) Both hepatic enzyme systems may be vulnerable to hypoxic–ischemic insult. (6) Maturational changes in medication metabolism can have a major clinical significance. Careful monitoring of serum levels of some medication classes (e.g., anticonvulsants) is necessary in the first weeks of life. D. Principles of medication excretion. b. Lungs: the lungs are an important route of excretion of gaseous anesthetics but are relatively less important for other medications. Excretion by the lungs is not well studied in the neonatal population. Because lung disease is common in newborn infants, adult data indicate that this may affect or limit the ability to excrete medications by this method. c. GI tract: the large, lipid-soluble surface of the GI tract allows diffusion of medications into the bloodstream. The limited motility in neonates affects excretion and increases the potential for reabsorption of medications or metabolites back into the circulation. d. Liver: the most important site of medication biotransformation also serves as an important site of medication excretion. The excretion of bile is an important means of medication elimination. (2) Metabolite or medication elimination in bile is dependent on the solubility characteristics of that substance in bile. e. Kidneys: the most important site for medication excretion. (b) As a percentage of cardiac output, renal blood flow is limited in neonates in comparison with older children and adults. Limited renal blood flow as an absolute value and as a percentage of cardiac output restricts medication or metabolite delivery to the kidney for excretion. (c) Renal blood flow increases with increasing gestational and postnatal age. (d) High umbilical artery catheter placement may reduce the renal blood flow. (2) Glomerular filtration rate (GFR): the removal, by passive filtration, of small unbound medication molecules at the glomerulus. Glomerular filtration is dependent on renal blood flow, the characteristics of the glomerular membranes, and the water solubility of the medication. (a) The GFR function is extremely limited in neonates (full-term neonates have 30% of adult values, per unit of body surface area; adult values are reached by 8 to 12 months of age) (Stewart and Hampton, 1987; van den Anker et al., 2011). (b) GFR is related to gestational age; the lower the gestational age, the lower the GFR. Neonates born at less than 34 weeks of gestation have fewer glomeruli, with total glomerular mass proportional to gestational age (Stewart and Hampton, 1987). Glomerular filtration has been shown to mature rapidly the first 2 weeks of life, although GFR in premature neonates remains significantly suppressed as compared to full-term neonates (Funk et al., 2012). (c) GFR can be further compromised with asphyxia, hypoxia, or indomethacin treatment. Limited glomerular filtration reduces removal of medications or metabolites at the glomerulus. (d) Medication excretion dependent on glomerular filtration includes indomethacin, digoxin, aminoglycosides, and vancomycin. (3) Tubular secretion: the active secretion of molecules into the tubular urine. Tubular secretion is dependent on the efficiency of tubular function. (a) Neonates have a relatively small mass of functional tubular cells, as well as an immaturity of tubular function. This functional limitation is thought to be caused in part by a decrease of renal blood flow to the renal tubular region and by shortened renal tubules (Stewart and Hampton, 1987). (b) The limitation in tubular mass and function causes poor excretion of medications and metabolites removed by this method. (c) Tubular secretion matures much more slowly than glomerular filtration. (d) Tubular secretion is also vulnerable to hypoxic–ischemic insult. (e) Medications dependent on tubular secretion for excretion include penicillins, morphine, and thiazide diuretics. (4) Tubular reabsorption: reabsorption, for some medications, back into the circulating plasma. (a) May occur through either passive diffusion or active transport. (b) Passive diffusion appears to be the most important process. (c) Tubular reabsorption matures much more slowly than glomerular filtration. (d) Substances dependent on tubular reabsorption include caffeine, glucose, phosphate, and sodium. (5) Urinary output: (b) Blood level monitoring is required to ensure safe serum levels of renally excreted medications. B. Definitions. b. Bactericidal medications: agents that kill microorganisms. At lower concentrations they may be bacteriostatic (e.g., aminoglycosides, penicillin, cephalosporins). 2. Minimal inhibitory concentration (MIC): the lowest concentration of a medication that stops visible organism growth in a laboratory setting. In the body, this cannot be directly measured and is dependent on achieved tissue concentration and bacterial count. This is easily measured in the microbiology laboratory and is used to measure the susceptibility of the microorganism to antimicrobial agents. 3. Minimal bactericidal concentration (MBC): the lowest concentration of a medication that results in a ≥ 99.9% decline in microbial number, measured in the laboratory setting. It is useful when compared with known potential toxic concentration levels to choose the antimicrobial regimen that does the greatest good with the fewest adverse or toxic effects. 4. Resistance: the ability of microorganisms to counteract the bacteriostatic or bactericidal effects of an antimicrobial agent. b. Microorganisms develop resistance by: (2) Decreased cellular penetration: microorganisms alter their cell wall permeability to prevent penetration of antimicrobial agents into the cell (e.g., Pseudomonas aeruginosa alters porin channels to limit the entry of ceftazidime, imipenem–cilastatin). (3) Altered target proteins: microorganisms change target proteins so that antimicrobial agents cannot bind and elicit antimicrobial activity (e.g., Streptococcus pneumoniae’s resistance to penicillin). (4) Efflux pump: microorganisms pump the antimicrobial agent out of the cell before the antimicrobial agent can kill the microorganism (e.g., Streptococcus pneumoniae’s resistance to erythromycin). C. Basic principles of antimicrobial use. b. Must be readily achievable and sustainable for the desired duration of antimicrobial therapy. 2. The choice of antimicrobial agent(s) must be taken into account: a. Microorganism susceptibility to available antimicrobial agents. b. Relative permeability of the target tissue to agent of choice (e.g., blood–brain barrier). c. Bioactivity of chosen antimicrobial agent in target tissue (e.g., bactericidal or bacteriostatic). d. Known MIC/MBC in relation to existing body of knowledge concerning side effects and toxic effects in the specific population. e. Specific characteristics of the individual patient in relation to the chosen antimicrobial’s toxicities (e.g., the blood levels of a nephrotoxic antimicrobial such as gentamicin should be closely monitored in patients with impaired renal function). D. Specific considerations in the neonatal population. b. Differences in response or potential for toxic effects may result from immaturity and/or illness state. 2. Pharmacokinetics. b. Absorption. (2) Changes in skin permeability in the extremely immature neonate may allow topically applied antimicrobial agents to be absorbed systemically. (3) Blood flow changes may affect absorption and distribution of antimicrobials administered intramuscularly or subcutaneously (e.g., repeated intramuscular administration of aminoglycosides to premature neonates may result in local tissue damage and unacceptably variable rates of absorption). c. Distribution: Decreasing body water and increasing body fat concentration in more mature neonates affect the volume through which the antimicrobial agent is distributed. This may make dosage adjustments necessary in the first days of life. d. Metabolism: Limited hepatic function, because of immaturity or illness state, may affect dosage regimen of some antibiotics, requiring smaller or less frequent doses of some antibiotics (e.g., nafcillin, erythromycin). e. Excretion: (2) Limited renal function or failure may cause toxic effects as a result of accumulation of medication or metabolites (e.g., nephrotoxicity from aminoglycosides). (3) Medication may have a direct effect on renal function. (a) Blood flow to kidney may be diminished (e.g., amphotericin can cause renal artery vasospasms). 2. The use of cardiovascular agents is increasing in the care of neonates. B. Basic principles of cardiovascular medication use. 2. Knowledge of the pathophysiologic basis of neonatal cardiovascular disease is necessary to ensure proper application of this class of medications. 3. Many of these medications have overlapping or synergistic effects. This overlap in clinical response makes the optimal choice of a medication and dose difficult. 4. Extensive knowledge and application of invasive and noninvasive cardiovascular monitoring techniques in the neonatal population are necessary to allow titration of the medication dose to the clinical response. C. Types of cardiovascular medications. 1. Inotropic/vasopressive agents. b. Used both for cardiovascular resuscitation and long-term support of the myocardium. c. Specific inotropic agents: (2) Sympathomimetic amines (e.g., epinephrine, dopamine, dobutamine, isoproterenol). ii. α2-adrenergic receptor response: activation of central nervous system (CNS) receptors in the brain to suppress outflow of the sympathetic nervous system activity from the brain. Results in decreased motility and tone of intestine and stomach. iii. β1-adrenergic receptor response: increased strength and rate of myocardial contraction. iv. β2-adrenergic receptor response: vascular smooth muscle dilation and bronchial muscle relaxation. v. Five different dopamine receptors are located in multiple organs and have mixed actions involving motor function, cardiovascular (e.g., vasodilating properties), renal (e.g., intrarenal vasodilator), behavioral, and hormonal (e.g., prolactin secretion) systems.For example, epinephrine increases blood pressure by stimulating the α1– and β1– receptors. Dobutamine increases cardiac output by stimulating the β1-receptors. (b) Response to each of these medications depends on relative amounts of α-, β-, and dopamine receptor effects. (c) Prolonged administration of sympathomimetic amines may result in diminished clinical efficacy secondary to diminished responses of α- and β-receptors, referred to as tachyphylaxis. 2. Antihypertensives/vasodilators. b. May be used to inhibit pathophysiologic changes that cause increased blood pressure (e.g., captopril, propranolol) or directly reduce blood pressure through changes in intravascular volume (e.g., diuretics) or vascular resistance (e.g., hydralazine). 3. Antiarrhythmics. a. Used to treat cardiac dysrhythmias causing adverse effects on cardiovascular stability. b. Include adenosine, digoxin, esmolol, and lidocaine. D. Specific considerations in the neonatal population. b. Cardiovascular medications are commonly used in conjunction with other medications that may affect the neonate’s response to the medication regimen (e.g., the digoxin–furosemide combination may result in electrolyte loss with the diuretic, which in turn may potentiate a toxic response to the digoxin). 2. Pharmacokinetics. b. Distribution. (1) Poor cardiac output/shock states may affect the distribution of medication to all tissues. (2) Medication administered for a desired response to one target organ may cause an undesirable systemic response (e.g., dobutamine increases cardiac output but may cause decreased blood pressure, furosemide reduces pulmonary edema but may decrease blood pressure). (3) Some cardiovascular medications are highly albumin-bound; this raises the possibility of unconjugated bilirubin displacement from albumin. c. Metabolism. (2) Medication metabolites may cause a toxic response (e.g., cyanide liberation as a result of nitroprusside metabolism). (3) Rapid metabolism and serum clearance may require continuous IV infusion (e.g., dopamine, dobutamine). d. Excretion: (2) Medication may have a direct effect on renal function. (a) Urinary output may be diminished (e.g., indomethacin reduces the glomerular filtration rate). 1. In adult patients, these are the most widely used group of medications. 2. The value of pain control and mood alteration in the neonatal population has only recently been recognized. 3. Recent increased interest in the use of CNS medications in the neonatal population has caused a recognition that the body of knowledge about these medications is limited. 4. The use of these medications is increasing as neurobehavioral assessment skills increase among neonatal caregivers. B. Definitions. 2. Anesthetic medication: a medication that removes pain sensation either through peripheral nerve block (e.g., lidocaine) or through CNS effects (e.g., high-dose fentanyl). Not all anesthetic medications provide pain relief (e.g., inhalation gases, propofol). 3. Sedative–hypnotic medications: medications that provide mood alteration in patients with anxiety. These are divided into two groups: barbiturates (e.g., phenobarbital) and nonbarbiturates (e.g., chloral hydrate, lorazepam). These medications do not provide relief from pain. 4. Addiction: a lifestyle change that occurs in a medication-dependent person. This lifestyle change involves a focus on medication use. This behavior cannot occur in a neonate. 5. Tolerance: a condition that may occur with many types of medications. Tolerance exists when larger doses and higher serum concentrations of the medication are required to achieve the desired response, and commonly occurs in conjunction with physical dependence. 6. Dependence: a physiologic state in which the individual requires regular medication administration for continued physiologic well-being. Patients who develop dependence on a medication may need a dosage-tapering regimen rather than abruptly discontinuing the medication in order to prevent withdrawal symptoms. In neonates, dependence can develop from either placental transfer of medications or iatrogenically due to long-term infusions of sedatives and/or narcotics. C. Basic principles of CNS medication use. 1. Mechanism of action of most CNS medications is not clearly understood. 2. Assessment of need for these medications must be carefully performed as an ongoing process. a. Close attention must be paid to differentiation of need for sedation, pain relief, or both. b. These medications may cause the development of medication tolerance and/or dependence. 3. Consideration must be made for the risks and benefits of the medication in relation to potential side effects or toxicities. 4. The science of the study of neonatal neurologic development is still emerging, and much is yet to be learned. The effect that CNS medications may have on that development is largely unknown. D. Specific considerations in the neonatal population. b. Specific physiologic characteristics in the neonatal population require careful observation for harmful side effects or toxicities (e.g., reduction in blood pressure with IV bolus doses of morphine, chest wall rigidity with fentanyl). c. Narcotic analgesics may cause respiratory depression and may precipitate respiratory failure in neonates. Therefore, the lowest possible dose that relieves the patient’s pain should be used. d. Careful assessment of clinical response is necessary to determine the most safe and effective dose and interval. 2. Pharmacokinetics. (2) Oral or rectal absorption of mild analgesics and sedatives is often adequate (e.g., acetaminophen). b. Distribution. (2) As neonates increase their proportion of body fat, the doses required will increase. (3) Because these agents are stored in the body fat, patients must be monitored for the accumulation of these agents and hence prolonged effects. c. Metabolism. (1) Slower hepatic metabolism may cause a prolonged half-life. (2) Hepatic disease may markedly increase the risk of toxic effects (e.g., chloral hydrate). (3) Hepatic metabolism may convert medications to either toxic metabolites or active metabolites (e.g., theophylline converts to caffeine). d. Excretion. 1. Commonly used in neonatal patients to promote the removal of excessive extracellular fluid. 2. Site of action of nearly all diuretic agents is the luminal surface of the renal tubular cell. B. Basic principles of diuretic use. 2. Diuretic medications whose primary purpose is to cause the excretion of excess extracellular fluid commonly cause a secondary or side effect of loss of electrolytes, along with the desired water loss. Knowledge of the specific action of each diuretic medication will assist the clinician in monitoring electrolytes for undesirable losses. 3. The pharmacologic response is dependent on the existing level of renal function and the medication’s ability to reach the target tissue in amounts adequate to produce the desired diuretic effect. 4. Any medication or therapy that increases the GFR may have an indirect diuretic effect. Some medications that act on the cardiovascular system to increase cardiac output or increase renal blood flow through vasodilation may cause diuresis (e.g., dopamine). Maximal water and electrolyte excretion usually occurs in the first days of use. Later, decreased GFR and hyperaldosteronism resulting from diuretic-induced hypovolemia limit these losses. C. Specific considerations in the neonatal population. b. The renal tubular function is limited in all neonates and is more limited in less mature neonates. c. The clinical response to diuretic agents is commonly decreased because of existing poor renal tubular absorption. Therefore, a larger dose is needed to achieve the response. d. Limited tubular function potentiates electrolyte loss with many diuretic agents (e.g., furosemide, hydrochlorothiazide). 2. Pharmacokinetics. b. Distribution. Some diuretic medications are strongly protein-bound. Some concern has been raised over the displacement of bilirubin from albumin-binding sites (e.g., furosemide, spironolactone). c. Metabolism. Some diuretics are primarily metabolized by the liver (e.g., spironolactone). d. Excretion. (2) Some diuretics are primarily eliminated unchanged in the urine (e.g., furosemide, hydrochlorothiazide). 1. Vaccines are commonly given to the hospitalized neonatal patient. 2. Each year in January, the Centers for Disease Control and Prevention (CDC) Advisory Committee on Immunization Practices publishes an updated version of the recommended immunization guidelines. Each nurse should be familiar with these guidelines. 3. The following documentation is required when vaccines are given: a. Name, title, and business address of person administering the vaccine. b. Vaccine administered. c. Manufacturer of the vaccine. d. Lot number of the vaccine. e. Expiration date of vaccine. f. Date of administration. g. Site of administration. h. Route of administration. i. Documentation that the vaccine information statement (VIS) has been given to the parents/guardians. j. Documentation of the VIS publication date (the most current version must be used). B. Basic principles of immunization use. 1. The goal of immunization is to prevent many viral and bacterial diseases and their sequelae. 2. For maximum effectiveness, the immunizations must be given at specific ages before the recipients have been exposed to the diseases. 3. Live-virus vaccines (e.g., measles-mumps-rubella [MMR], rotavirus, varicella) should NOT be administered in the NICU. 4. All adverse events associated with immunization should be reported in detail in the patient’s medical record and also on the Vaccine Adverse Event Reporting System (VAERS) form found on the U.S. Food and Drug Administration (FDA) website. C. Types of immunizations. b. Some immunizing agents provide complete protection against disease for life (e.g., polio vaccine), some provide partial protection (e.g., influenza vaccine), and some must be readministered at intervals (booster doses) (e.g., tetanus vaccine). c. Vaccines may be either live (attenuated) or killed (inactivated). Inactivated vaccines may not elicit the range of immunologic response provided by attenuated agents. 2. Passive immunization. a. Involves the administration of preformed antibody to a recipient. b. Most often used when a person exposed to a disease has a high likelihood of complications from that disease and time does not permit adequate protection by active immunization alone. c. Can be used when a disease is already present with the hope of reducing the reaction to the disease. d. Accomplished through the use of immune globulin preparations (e.g., hepatitis B immune globulin, palivizumab). e. There are strict indications for the use of these products (American Academy of Pediatrics [AAP], 2012). D. Specific considerations in the neonatal population. b. Preterm infants who are medically stable should receive the immunizations at the recommended chronologic age as long as the following criteria are met: (1) The infant does not require ongoing treatment for serious infections or metabolic disease. (2) The infant does not have acute renal, cardiovascular, or respiratory illness. (3) The infant demonstrates a clinical course of sustained recovery and pattern of steady growth (AAP, 2012). c. The dose should NOT be decreased in preterm infants. d. Antibody titers may be measured to prove adequate immune responses. 2. Pharmacokinetics. (2) The preferred site for administration is the anterolateral aspect of the upper thigh. (3) The deltoid area of the upper arm can be used in older infants (> 1 year). (4) The upper, outer aspect of the buttocks should not be routinely used because of the possibility of damaging the sciatic nerve and reduced immunogenicity. (5) When necessary, two vaccines can be given in the same limb but should be separated by at least 1 inch if possible so that local reactions are not likely to overlap. (6) The recommended routes of administration are included in the package inserts of the vaccines (e.g., intramuscular, subcutaneous). b. Distribution. Vaccines should be held when the neonate is on pressor agents because the blood flow to the muscle and subcutaneous tissues may be limited. c. Metabolism/Excretion: Kidney and/or liver failure should not prevent the administration of vaccines. E. Misconceptions about vaccine contraindications: The nurse should be familiar with the specific contraindications listed in the package inserts of the vaccines so that immunizations are not withheld unnecessarily (AAP, 2012). There are several areas in which the nursing staff are an invaluable resource for the patient in helping to prevent errors in this fragile patient population. B. Standard concentrations for IV continuous infusions should be used rather than relying on concentrations based on weight. C. Double check of medication doses involves recalculating the weight-based doses and comparing prescribed doses with current medication dosing reference books to prevent potential medication dosing errors. In addition, smart pump technology that includes dosing ranges for both intermittent and continuous infusions for IV medications should be used with IV pumps to avoid programmable errors. D. Cross-checking on a regular basis is a nursing responsibility. Because of the very small volumes of medications commonly given to the NICU patient, a system for regular cross-checking of medications’ volume accuracy with medication concentration before administration should be established for all medications and especially with high-risk medications. E. Monitoring renal function through intake and output measurements may alert the care team to potential changes in medication metabolism and/or excretion. F. Meticulous medication administration and documentation involves administering the medication at the correct time and at the correct time interval. In addition, documenting the medication administration date, time, dose, route, and interval and the time medication levels are obtained is important in interpreting patient response to therapy. G. Facilitation of medication serum level monitoring with absolute accuracy makes safe administration of medications—with a narrow margin between effective and toxic levels—possible. H. Careful assessment of vital sign parameters and clinical responses may assist in the evaluation of desirable or undesirable medication responses. I. Careful observation of the neonate for therapeutic and toxic medication effects will allow medication administration to achieve the maximal desired response while minimizing toxic responses. J. Medication precautions must be observed. Any medication or medication preparation known to have a high risk of adverse effects in the neonate should be removed from the patient care area (e.g., concentrated potassium vials) or should be specifically labeled to avoid inadvertent administration. K. Facilitation of or participation in clinical trials designed to evaluate a medication’s efficacy does much to advance the body of knowledge related to neonatal pharmacology. L. Recognition of established clinical experience with individual medications in the neonatal population is essential. Because some medications are introduced into the clinical area after only minimal study of specific medication response in the neonate, early observation of potential toxic effects may avert a later disaster.
Pharmacology
PRINCIPLES OF PHARMACOLOGY
Terminology
PHARMACODYNAMICS
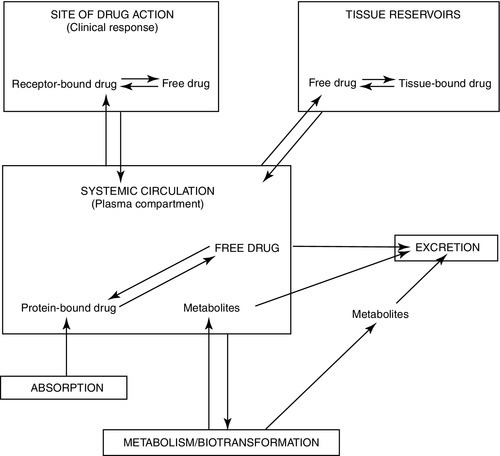
PHARMACOKINETICS
MEDICATION CATEGORIES
Antimicrobial Agents
Cardiovascular Agents
Central Nervous System Medications
Diuretics
Immunizations
NURSING IMPLICATIONS FOR MEDICATION ADMINISTRATION IN THE NEONATE
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


