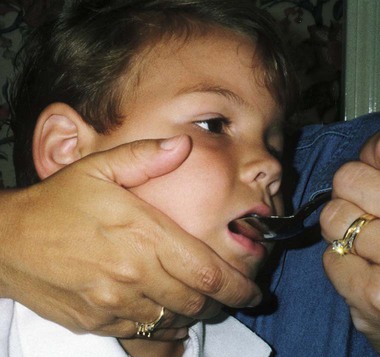
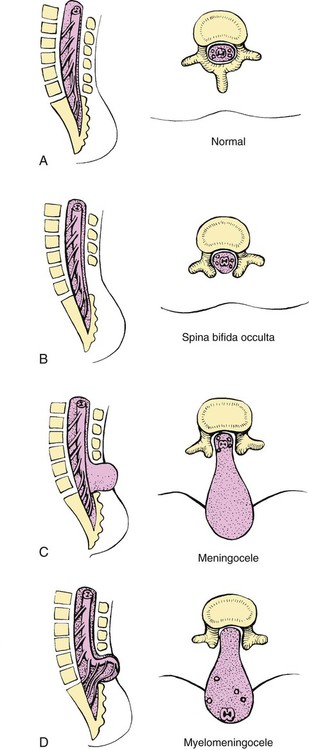
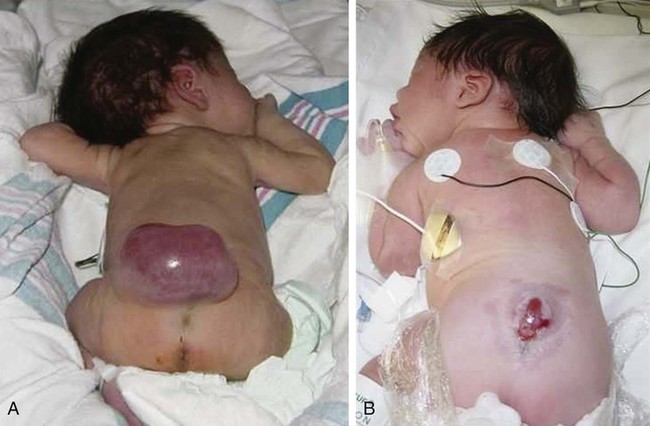
On completion of this chapter the reader will be able to: • Discuss the nursing role in helping parents care for the child who has cerebral palsy. • Formulate a nursing care plan for the preoperative and postoperative care of a child with myelomeningocele. • Outline a care plan for a child with Guillain-Barré syndrome. • Discuss the home care management of the child with a neuromuscular disease such as spinal muscular atrophy. • Discuss the prevention and treatment of tetanus. • Identify the causes of botulism in infants and children. http://evolve.elsevier.com/wong/essentials Animation—Guillain-Barré Syndrome Nursing Care Plans—The Child with Cerebral Palsy; The Child with Myelomeningocele (Spina Bifida) A new definition proposed in 2006 describes cerebral palsy (CP) as a “group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain” (Rosenbaum, Paneth, Leviton, and others, 2007). In addition to motor disorders, the condition often involves disturbances of sensation, perception, communication, cognition, and behavior; secondary musculoskeletal problems; and epilepsy (Rosenbaum, Paneth, Leviton, and others, 2007). The etiology, clinical features, and course vary and are characterized by abnormal muscle tone and coordination as the primary disturbances. CP is the most common permanent physical disability of childhood, and the incidence is reported to be between 2.4 to 3.6 per every 1000 live births in the United States (Hirtz, Thurman, Gwinn-Hardy, and others, 2007; Yeargin-Allsopp, Van Naarden Braun, Doernberg, and others, 2008). Since the 1960s, the prevalence of CP has risen approximately 20%, which most likely reflects the improved survival of extremely low–birth-weight and very low–birth-weight infants. Although the prevalent traditional hypothesis has been that CP results from perinatal problems, especially birth asphyxia, it is now believed that CP results more often from existing prenatal brain abnormalities; the exact cause of these abnormalities remains elusive but may include genetic factors, including clotting disorders as well as brain malformations. It has been estimated that as many as 80% of CP cases are attributable to unidentified prenatal factors (Krigger, 2006). Intrauterine exposure to maternal chorioamnionitis is associated with an increased risk of CP in infants of normal birth weight and preterm infants (Hermansen and Hermansen, 2006); however, not all term infants exposed to chorioamnionitis develop CP (Grether, Nelson, Walsh, and others, 2003; Wu, Escobar, Grether, and others, 2003). Perinatal ischemic stroke is also associated with a later diagnosis of CP (Golomb, Saha, Garg, and others, 2007). Additional factors that may contribute to the development of CP postnatally include bacterial meningitis, multiple births, viral encephalitis, motor vehicle crashes, and child abuse (shaken baby syndrome [traumatic brain injury]) (Krigger, 2006). One study found a higher risk of CP occurring among infants born at 42 weeks or later than among those born at 37 or 38 weeks’ gestation (Moster, Wilcox, Vollset, and others, 2010). A significant percentage (15% to 60%) of children with CP also have epilepsy. In summary, as many as 80% of the total cases of CP may be linked to a perinatal or neonatal brain lesion or brain maldevelopment, regardless of the cause (Krageloh-Mann and Cans, 2009). There are a few exceptions. In some cases, the manifestations or etiology are related to anatomic areas. For example, CP associated with preterm birth is usually spastic diplegia caused by hypoxic infarction or hemorrhage with periventricular leukomalacia in the area adjacent to the lateral ventricles. The athetoid (extrapyramidal) type of CP is most likely to be associated with birth asphyxia but can also be caused by kernicterus and metabolic genetic disorders such as mitochondrial disorders and glutaricaciduria (Johnston, 2011). Hemiplegic (hemiparetic) CP is often associated with a focal cerebral infarction (stroke) secondary to an intrauterine or perinatal thromboembolism, usually a result of maternal thrombosis or hereditary clotting disorder (Johnston, 2011). Cerebral hypoplasia and sometimes severe neonatal hypoglycemia are related to ataxic CP. Generalized cortical and cerebral atrophy often cause severe quadriparesis with cognitive impairment and microcephaly. A revision of the Winter classification was proposed in 2005 to reflect the child’s actual clinical problems and their severity, an assessment of the child’s physical and quality-of-life status across time, and long-term support needs (Bax, Goldstein, Rosenbaum, and others, 2005; Nehring, 2010). The proposed new definition has four major dimensions of classification (Bax, Goldstein, Rosenbaum, and others, 2005): Motor abnormalities—Nature and typology of the motor disorder; functional motor abilities Associated impairments—Seizures; hearing or vision impairment; attentional, behavioral, communicative, or cognitive deficits; oral motor and speech function Anatomic and radiologic findings—Anatomic distribution or parts of the body affected by motor impairments or limitations; radiologic findings sometimes including white matter lesions or brain anomaly noted on computed tomography (CT) or magnetic resonance imaging (MRI) Causation and timing—Identification of a clearly identified cause such as a postnatal event (e.g., meningitis, traumatic brain injury) Cerebral palsy has four primary types of movement disorders: spastic, dyskinetic, ataxic, and mixed (Nehring, 2010). The most common clinical type, spastic CP, represents an upper motor neuron muscular weakness (Box 32-1). The reflex arc is intact, and the characteristic physical signs are increased stretch reflexes, increased muscle tone, and (often) weakness. Early neurologic manifestations are usually generalized hypotonia or decreased tone that lasts for a few weeks or may extend for months or even as long as 1 year. Early recognition is made more difficult by the lack of reliable neonatal neurologic signs. However, nurses should monitor infants with known etiologic risk factors and evaluate them closely in the first 2 years of life. Because cortical control of movement does not occur until later in infancy, motor impairment associated with voluntary control is usually not apparent until after 2 to 4 months of age at the earliest. More often the diagnosis cannot be confirmed until the age of 2 years because motor tone abnormalities may be indicative of another neuromuscular illness. In addition, some children who show signs consistent with CP before 2 years do not demonstrate such signs after 2 years (Nehring, 2010). However, there is no consensus regarding an age cut-off for the onset of symptoms. Clinical manifestations of CP at the time of diagnosis are listed in Box 32-2; early warning signs are listed in Box 32-3, but these are not considered diagnostic. A number of assessment instruments are now available to evaluate muscle spasticity; functional independence in self-care, mobility, and cognition; self-initiated movements over time; and capability and performance of functional activities in self-care, mobility, and social function (Krigger, 2006). 1. To establish locomotion, communication, and self-help skills 2. To gain optimal appearance and integration of motor functions 3. To correct associated defects as effectively as possible 4. To provide educational opportunities adapted to the child’s needs and capabilities 5. To promote socialization experiences with other affected and unaffected children Ankle–foot orthoses (AFOs, braces) are worn by many of these children and are used to help prevent or reduce deformity, increase the energy efficiency of gait, and control alignment. Wheeled go-carts that provide sitting balance may serve as early “wheelchair” experience for young children. Manual or powered wheelchairs allow for more independent mobility (Figs. 32-1 and 32-2). Strollers can be equipped with custom seats for dependent mobilization. Botulinum toxin A (Botox) is also used to reduce spasticity in targeted muscles. Botulinum toxin A is injected into a selected muscle (commonly the quadriceps, gastrocnemius, or medial hamstrings) after a topical anesthetic is applied. The drug acts to inhibit the release of acetylcholine into a specific muscle group, thereby preventing muscle movement. When it is administered early in the course of the condition, affected muscle contractures may be minimized, particularly in lower extremities, thus avoiding surgical procedures with possible adverse effects. The goal is to allow stretching of the muscle as it relaxes and permit ambulation with an AFO. The major reported adverse effects of botulinum toxin A injection are pain at the injection site and temporary weakness (Lukban, Rosales, and Dressler, 2009). Prime candidates for botulinum toxin A injections are children with spasticity confined to the lower extremities; the drug weakens spasticity so the muscles can be stretched and the child may walk with or without orthoses. The onset of action occurs within 24 to 72 hours, with a peak effect observed at 2 weeks and a duration of action of 3 to 6 months. Children with CP may also experience pain as a result of surgical procedures intended to reduce contracture deformities, body position, gastroesophageal reflux, and physical therapy (McKearnan, Kieckhefer, Engel, and others, 2004). Therefore, pain management is an important aspect of care of children with CP. Antiepileptic drugs (AEDs) such as carbamazepine (Tegretol) and divalproex (valproate sodium and valproic acid; Depakote) are prescribed routinely for children who have seizures. Other medications include levodopa to treat dystonia; Artane for treating dystonia, and for increasing the use of upper extremities and vocalizations; and reserpine for hyperkinetic movement disorders such as chorea or athetosis (Johnston, 2011). All medications should be monitored for maintenance of therapeutic levels and avoidance of subtherapeutic or toxic levels. There is some evidence that neuromuscular electrical stimulation (NMES) in addition to dynamic splinting may result in increased muscle strength, range of motion, and function of upper limbs in children with CP. Further studies are needed in children with CP to support the use of botulinum toxin A in conjuction with NMES to decrease muscle spasticity and improve function (Wright, Durham, Ewins, and others, 2012). Behavior problems may occur and often interfere with the child’s development. Attention-deficit/hyperactivity disorder and other learning problems require professional attention. In addition, children with CP may have vision difficulties such as strabismus, nystagmus, and optic atrophy (Johnston, 2011). Speech-language therapy involves the services of a speech-language pathologist who may also assist with feeding problems. The prognosis for the child with CP depends largely on the type and severity of the condition. Children with mild to moderate involvement (85%) have the capability of achieving ambulation between the ages of 2 and 7 years (Berker and Yalçin, 2008). If the child does not achieve independent ambulation by this time, chances are poor for ambulation and independence. Approximately 30% to 50% of individuals with CP have significant cognitive impairments, and an even higher percentage have mild cognitive and learning deficits. However, many children with severe spastic tetraplegic CP have normal intelligence. Growth is affected in children with spastic tetraplegia, and many children remain below the 5th percentile for age and sex. As children with CP become adults, about 30% remain in the home and are cared for by a parent or caregiver; 50% of individuals with spastic tetraplegia live in independent settings and function at appropriate social levels considering their disability (Green, Greenberg, and Hurwitz, 2003). Vocational rehabilitation and higher education are possible for adults with CP. Children with severe CP mobility impairment and feeding problems often succumb to respiratory tract infection in childhood. The few survival rate studies on children or adults with CP show that survival is influenced by existing comorbidities (Nehring, 2010). Prevention of some cases of CP may become a reality in the near future. Studies indicate that early neuroprotection in term infants with the use of therapeutic hypothermia (head cooling or whole-body cooling) within 6 hours of birth improved survival without CP by approximately 40% (Johnston, Fatemi, Wilson, and others, 2011). The Child with Cerebral Palsy Because children with CP expend so much energy in their efforts to accomplish ADLs, more frequent rest periods should be arranged to avoid fatigue. The diet should be tailored to the child’s activity and metabolic needs. Gastrostomy feedings may be necessary to supplement regular feedings and ensure adequate weight gain, particularly in children at risk for growth failure and chronic malnutrition, those with severe CP and subsequent oral feeding difficulties, and children whose well-being is affected by illness and decreased fluid or medication intake (Rogers, 2004). Oral feedings may be continued to maintain oral motor skills. Weight gain is perceived as an important measure of adequate oral feeding efficiency. Parents may need assistance and advice with medication administration through a gastrostomy tube to prevent clogging. A skin-level gastrostomy is particularly suited for children with CP. Because jaw control is often compromised, more normal control can be achieved if the feeder provides stability of the oral mechanism from the side or front of the face. When directed from the front, the middle finger of the nonfeeding hand is placed posterior to the body portion of the chin, the thumb is placed below the bottom lip, and the index finger is placed parallel to the child’s mandible (Fig. 32-3). Manual jaw control from the side assists with head control, correction of neck and trunk hyperextension, and jaw stabilization. The middle finger of the nonfeeding hand is placed posterior to the bony portion of the chin, the index finger is placed on the chin below the lower lip, and the thumb is placed obliquely across the cheek to provide lateral jaw stability (Fig. 32-4). Safety precautions are implemented, such as having children wear protective helmets if they are subject to falls or capable of injuring their heads on hard objects. Because children with CP are at risk for altered proprioception and subsequent falls, the home and play environments should be adapted to their needs to prevent bodily harm. Appropriate immunizations should be administered to prevent childhood illnesses and protect against respiratory tract infections such as influenza or pneumonia. Dental problems may be more common in children with CP, which creates a need for meticulous attention to all aspects of dental care. Transportation of the child with motor problems and restricted mobility may be especially challenging for the family and child. Attention must be given to the child’s safety when riding in a motor vehicle; a federally approved safety restraint should be used at all times. It is recommended that children with CP ride in a rear-facing position as long as possible because of their poor head, neck, and trunk control (Lovette, 2008). Car restraints especially designated for children with poor head and neck control are available and should be used.* Probably the nursing interventions most valuable to the family are support and help in coping with the emotional aspects of the disorder, many of which are discussed in relation to the child with a disability (see Chapter 18). Initially, the parents need supportive counseling directed toward understanding the meaning of the diagnosis and all of the feelings that it engenders. Later they need clarification regarding what they can expect from the child and from health professionals. Educating families in the principles of family-centered care and parent–professional collaboration is essential. The family may require help in modifying the home environment for care of the child (see also Chapter 20). Transportation to the practitioner’s office and other health care agencies often requires special arrangements. Parents may also find help and comfort from parent groups, with whom they can share problems and concerns and from whom they can derive comfort and practical information. Parent support groups are most helpful through sharing experiences and accomplishments. For example, parents can learn from others what it is like to have a child with CP, which is generally not possible from professionals (see Family-Centered Care box). The national organization United Cerebral Palsy* has branches in most communities. The association provides a variety of services for children and families. A number of excellent books also are available to guide parents and nurses who work with children with CP. The Reality of Acceptance of Cerebral Palsy Acceptance is rarely achieved in the length of time implied in the literature. The bitterness is gone; I am now happy for people who have children without CP. He is now a young man of 25 years, and I am learning to accept his independence. Shriners Hospitals for Children Abnormalities that derive from the embryonic neural tube (neural tube defects [NTDs]) constitute the largest group of congenital anomalies that are consistent with multifactorial inheritance. Normally, the spinal cord and cauda equina are encased in a protective sheath of bone and meninges (Fig. 32-5, A). Failure of neural tube closure produces defects of varying degrees (Box 32-4). They may involve the entire length of the neural tube or may be restricted to a small area. In the United States, rates of NTDs have declined from 1.3 per 1000 births in 1970 to 0.3 per 1000 births after the introduction of mandatory food fortification with folic acid in 1998. One concern is that NTD rates have not decreased among Hispanic and non-Hispanic white mothers since 1999 (Centers for Disease Control and Prevention [CDC], 2009). In 2005, the rates for spina bifida (SB) were estimated by the CDC to be 17.96 per 100,000 live births, thus making this one of the most common birth defects in the United States (Matthews, 2009; Wolff, Witkop, Miller, and others, 2009). Increased use of prenatal diagnostic techniques and termination of pregnancies have also affected the overall incidence of NTDs. (see also Prevention, p. 1102). Spina bifida occulta refers to a defect that is not visible externally. It occurs most frequently in the lumbosacral area (L5 and S1) (Fig. 32-5, B). SB occulta may not be apparent unless there are associated cutaneous manifestations or neuromuscular disturbances. Spina bifida cystica refers to a visible defect with an external saclike protrusion. The two major forms of SB cystica are meningocele, which encases meninges and spinal fluid but no neural elements (Fig. 32-5, C), and myelomeningocele (or meningomyelocele), which contains meninges, spinal fluid, and nerves (Fig. 32-5, D). Meningocele is not associated with neurologic deficit, which occurs in varying, often serious, degrees in myelomeningocele. Clinically, the term spina bifida is used to refer to myelomeningocele. There is evidence of a multifactorial etiology, including drugs, radiation, maternal malnutrition, chemicals, and possibly a genetic mutation in folate pathways in some cases, which may result in abnormal development. There is also evidence of a genetic component in the development of SB; myelomeningocele may occur in association with syndromes such as trisomy 18, PHAVER (limb pterygia, congenital heart anomalies, vertebral defects, ear anomalies, and radial defects) syndrome, and Meckel-Gruber syndrome (Shaer, Chescheir, and Schulkin, 2007). Additional factors predisposing children to an increased risk of NTDs include prepregnancy maternal obesity, maternal diabetes mellitus, low maternal vitamin B12 status, maternal hyperthermia, and the use of AEDs in pregnancy. The genetic predisposition is supported by evidence of the risk of recurrence after one affected child (3%–4%) and a 10% risk of recurrence with two previously affected children (Kinsman and Johnston, 2011). The degree of neurologic dysfunction depends on where the sac protrudes through the vertebrae, the anatomic level of the defect, and the amount of nerve tissue involved. The majority of myelomeningoceles (75%) involve the lumbar or lumbosacral area (Fig. 32-6). Hydrocephalus is a frequently associated anomaly in 80% to 90% of the children. About 80% of patients with myelomeningocele develop a type II Chiari malformation (Kinsman and Johnston, 2011). The diagnosis of SB is made on the basis of clinical manifestations (Box 32-5) and examination of the meningeal sac. Diagnostic measures used to evaluate the brain and spinal cord include MRI, ultrasonography, and CT. A neurologic evaluation will determine the extent of involvement of bowel and bladder function as well as lower extremity neuromuscular involvement. Flaccid paralysis of the lower extremities is a common finding with absent deep tendon reflexes. It is possible to determine the presence of some major open NTDs prenatally. Ultrasonographic scanning of the uterus and elevated maternal concentrations of α-fetoprotein (AFP, or MS-AFP), a fetal-specific γ-1-globulin, in amniotic fluid may indicate anencephaly or myelomeningocele. The optimum time for performing these diagnostic tests is between 16 and 18 weeks of gestation before AFP concentrations normally diminish and in sufficient time to permit a therapeutic abortion. It is recommended that such diagnostic procedures and genetic counseling be considered for all mothers who have borne an affected child, and testing is offered to all pregnant women (Kirkham, Harris, and Grzybowski, 2005). Chorionic villus sampling is also a method for prenatal diagnosis of NTDs; however, it carries certain risks (skeletal limb depletion) and is not recommended before 10 weeks of gestation. Many authorities believe that early closure, within the first 24 to 72 hours, offers the most favorable outcome. Surgical closure within the first 24 hours is recommended if the sac is leaking CSF (Kinsman and Johnston, 2011). Associated problems are assessed and managed by appropriate surgical and supportive measures. Shunt procedures provide relief from imminent or progressive hydrocephalus (see Chapter 28). When diagnosed, ventriculitis, meningitis, urinary tract infection, and pneumonia are treated with vigorous antibiotic therapy and supportive measures. Surgical intervention for Chiari II malformation is indicated only when the child is symptomatic (i.e., high-pitched crowing cry, stridor, respiratory difficulties, oral-motor difficulties, upper extremity spasticity). Early surgical closure of the myelomeningocele sac through fetal surgery has been evaluated in relation to prevention of injury to the exposed spinal cord tissue and the improvement of neurologic and urologic outcomes in the affected child. The Management of Myelomeningocele Study, a clinical trial supported by the National Institute of Health, found that prenatal surgery for myelomeningocele reduced the need for shunting (for hydrocephalus), evaluated at 12 months, and there was an improvement in mental and motor function scores at 30 months in the children who had prenatal surgery (compared with children who had postnatal surgery) (Adzick, Thom, Spong, and others, 2011). Outcome data for urologic and bowel function are not available at this time.
The Child with Neuromuscular or Muscular Dysfunction

Congenital Neuromuscular or Muscular Disorders
Cerebral Palsy
Pathophysiology
Clinical Classification
Diagnostic Evaluation
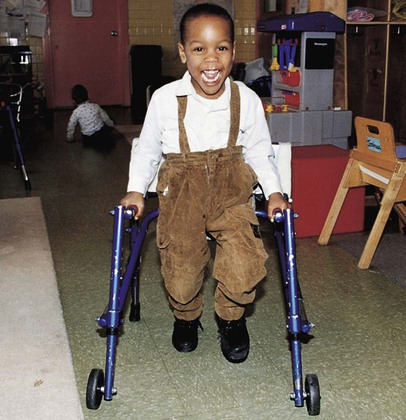
Therapeutic Management
Prognosis
Nursing Care Management
 Nursing Care Plan
Nursing Care Plan
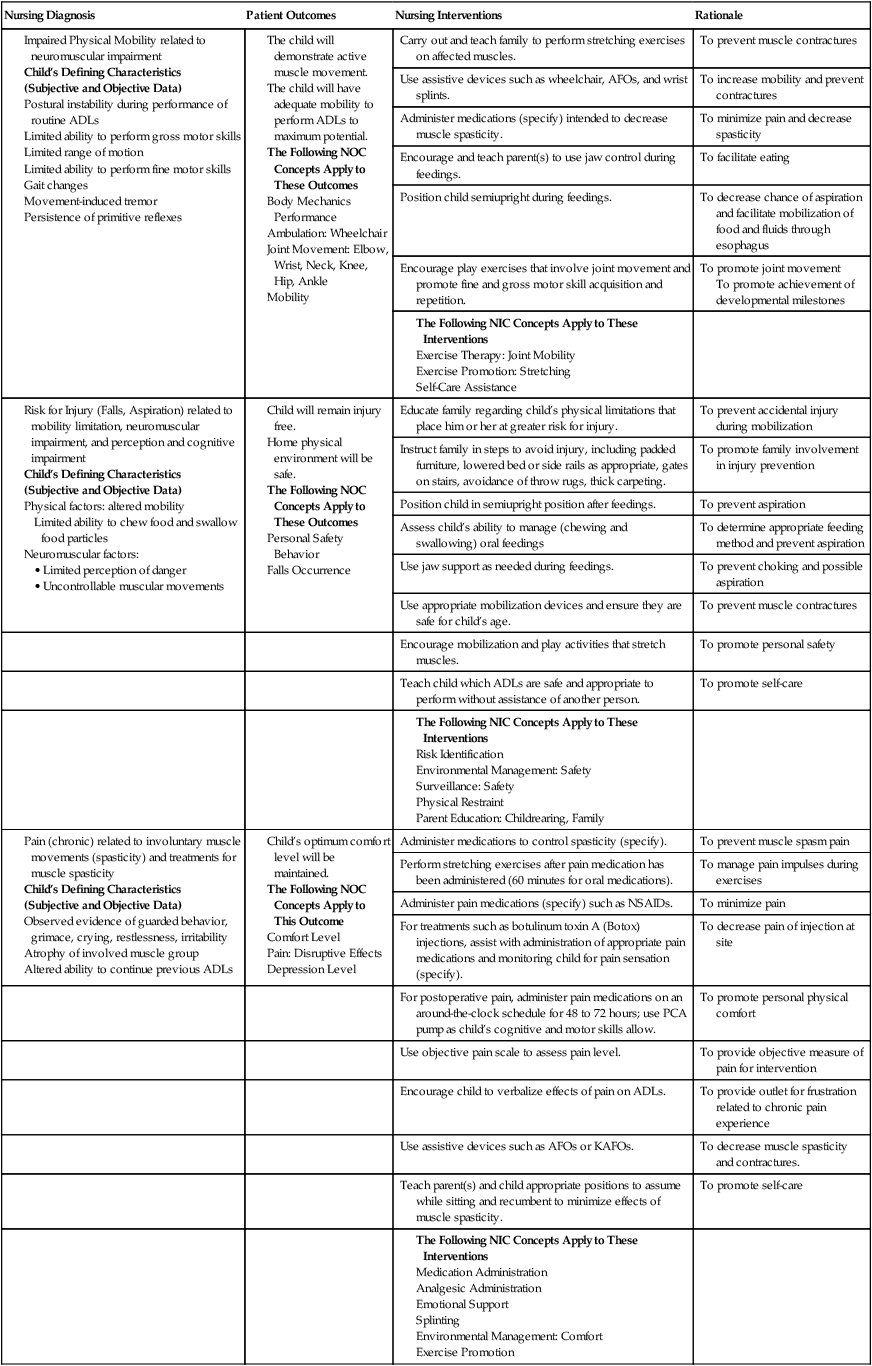
Nursing Diagnosis
Patient Outcomes
Nursing Interventions
Rationale
Carry out and teach family to perform stretching exercises on affected muscles.
To prevent muscle contractures
Use assistive devices such as wheelchair, AFOs, and wrist splints.
To increase mobility and prevent contractures
Administer medications (specify) intended to decrease muscle spasticity.
To minimize pain and decrease spasticity
Encourage and teach parent(s) to use jaw control during feedings.
To facilitate eating
Position child semiupright during feedings.
To decrease chance of aspiration and facilitate mobilization of food and fluids through esophagus
Encourage play exercises that involve joint movement and promote fine and gross motor skill acquisition and repetition.
To promote joint movement
To promote achievement of developmental milestones
Educate family regarding child’s physical limitations that place him or her at greater risk for injury.
To prevent accidental injury during mobilization
Instruct family in steps to avoid injury, including padded furniture, lowered bed or side rails as appropriate, gates on stairs, avoidance of throw rugs, thick carpeting.
To promote family involvement in injury prevention
Position child in semiupright position after feedings.
To prevent aspiration
Assess child’s ability to manage (chewing and swallowing) oral feedings
To determine appropriate feeding method and prevent aspiration
Use jaw support as needed during feedings.
To prevent choking and possible aspiration
Use appropriate mobilization devices and ensure they are safe for child’s age.
To prevent muscle contractures
Encourage mobilization and play activities that stretch muscles.
To promote personal safety
Teach child which ADLs are safe and appropriate to perform without assistance of another person.
To promote self-care
Administer medications to control spasticity (specify).
To prevent muscle spasm pain
Perform stretching exercises after pain medication has been administered (60 minutes for oral medications).
To manage pain impulses during exercises
Administer pain medications (specify) such as NSAIDs.
To minimize pain
For treatments such as botulinum toxin A (Botox) injections, assist with administration of appropriate pain medications and monitoring child for pain sensation (specify).
To decrease pain of injection at site
For postoperative pain, administer pain medications on an around-the-clock schedule for 48 to 72 hours; use PCA pump as child’s cognitive and motor skills allow.
To promote personal physical comfort
Use objective pain scale to assess pain level.
To provide objective measure of pain for intervention
Encourage child to verbalize effects of pain on ADLs.
To provide outlet for frustration related to chronic pain experience
Use assistive devices such as AFOs or KAFOs.
To decrease muscle spasticity and contractures.
Teach parent(s) and child appropriate positions to assume while sitting and recumbent to minimize effects of muscle spasticity.
To promote self-care
 Nursing Care Plan—The Child with Cerebral Palsy
Nursing Care Plan—The Child with Cerebral Palsy
Support the Family
 Family-Centered Care
Family-Centered Care
Neural Tube Defects (Myelomeningocele)
Etiology
Diagnostic Evaluation
Prenatal Detection
Therapeutic Management
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


The Child with Neuromuscular or Muscular Dysfunction
 Infants at risk according to known etiologic factors associated with CP warrant careful assessment during early infancy to identify the signs of neuromotor dysfunction as soon as possible. The neurologic examination and history are the primary means for diagnosis. Neuroimaging of the child with suspected brain abnormality and CP is now recommended for diagnostic assessment, with MRI preferred to CT scan. Metabolic and genetic testing is recommended if no structural abnormality is identified by neuroimaging; laboratory tests are no longer recommended in the diagnostic process for CP.
Infants at risk according to known etiologic factors associated with CP warrant careful assessment during early infancy to identify the signs of neuromotor dysfunction as soon as possible. The neurologic examination and history are the primary means for diagnosis. Neuroimaging of the child with suspected brain abnormality and CP is now recommended for diagnostic assessment, with MRI preferred to CT scan. Metabolic and genetic testing is recommended if no structural abnormality is identified by neuroimaging; laboratory tests are no longer recommended in the diagnostic process for CP. Case Study—Cerebral Palsy
Case Study—Cerebral Palsy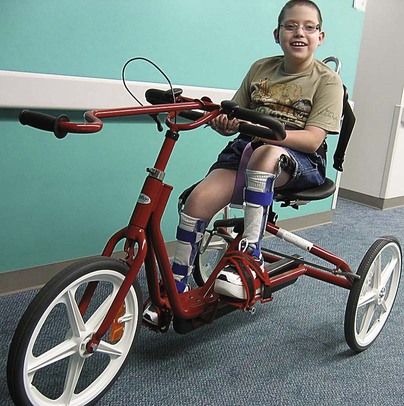
 Nursing Alert
Nursing Alert Because children with CP are being identified and treated at an earlier age, parents are participating earlier in treatment programs for their children with disabilities. They are taught the proper handling and home care of young children with CP and need a carefully planned program so that their change of role from parent to caregiver can be melded into the already established relationship. Close work with other multidisciplinary team members is essential. Nurses reinforce the therapeutic plan and assist the family in devising and modifying equipment and activities to continue the therapy program in the home. The nursing process in the care of the child with CP is outlined in the Nursing Care Plan.
Because children with CP are being identified and treated at an earlier age, parents are participating earlier in treatment programs for their children with disabilities. They are taught the proper handling and home care of young children with CP and need a carefully planned program so that their change of role from parent to caregiver can be melded into the already established relationship. Close work with other multidisciplinary team members is essential. Nurses reinforce the therapeutic plan and assist the family in devising and modifying equipment and activities to continue the therapy program in the home. The nursing process in the care of the child with CP is outlined in the Nursing Care Plan.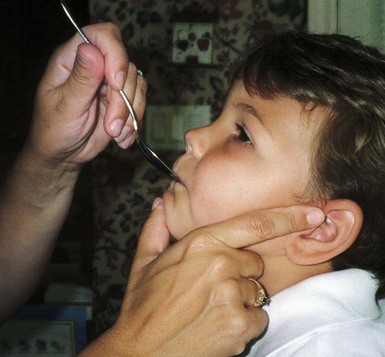
Get Clinical Tree app for offline access
