On completion of this chapter the reader will be able to: • Differentiate between the disorders caused by hypopituitary and hyperpituitary dysfunction. • Describe the manifestations of thyroid hypofunction and hyperfunction and the management of children with the disorders. • Distinguish between the manifestations of adrenal hypofunction and hyperfunction. • Differentiate among the various categories of diabetes mellitus. • Discuss the management and nursing care of the child with diabetes mellitus in the acute care setting. • Distinguish between a hypoglycemic and a hyperglycemic reaction. • Formulate a teaching plan for instructing the parents of a child with diabetes mellitus. http://evolve.elsevier.com/wong/essentials Animations—Adrenal Function; Insulin Injection Case Studies—Diabetes Mellitus; Diabetes Insipidus Nursing Care Plans—The Child with Diabetes Mellitus; The Child with Diabetic Ketoacidosis (DKA) The endocrine system consists of three components: (1) the cells, which send chemical messages by means of hormones; (2) the target cells, or end organs, which receive the chemical messages; and (3) the environment through which the chemicals are transported (blood, lymph, extracellular fluids) from the sites of synthesis to the sites of cellular action. The endocrine system controls or regulates metabolic processes governing energy production, growth, fluid and electrolyte balance, response to stress, and sexual reproduction (Baxter and Ribeiro, 2004). The endocrine glands, which are distributed throughout the body, are listed in Table 29-1; also listed are several additional structures sometimes considered endocrine glands, although they are not usually included. The pathophysiology review in Figure 29-1 provides a summary of the principle pituitary hormones and their target organs. TABLE 29-1 Promotes growth of bone and soft tissues Has main effect on linear growth Maintains a normal rate of protein synthesis Conserves carbohydrate utilization and promotes fat mobilization Is essential for proliferation of cartilage cells at epiphyseal plate Is ineffective for linear growth after epiphyseal closure *For each anterior pituitary hormone there is a corresponding hypothalamic-releasing factor. A deficiency in these factors caused by inhibiting anterior pituitary hormone synthesis produces the same effects. (See text for more detailed information.) †In males, LH is sometimes known as interstitial cell–stimulating hormone (ICSH). A hormone is a complex chemical substance produced and secreted into body fluids by a cell or group of cells that exerts a physiologic controlling effect on other cells (Kliegman, Stanton, St. Geme, and others, 2011). These effects may be local or distant and may affect either most cells of the body or specific “target” tissues. Hormones are released by the endocrine glands into the bloodstream, and production is regulated by a feedback mechanism (see Table 29-1). The master gland of the endocrine system is the anterior pituitary, which is in turn controlled by the hypothalamus. Some hormones, such as insulin, are regulated by other mechanisms. The pituitary gland is divided into two lobes, the anterior and the posterior lobes. Each lobe is responsible for different hormones. Disorders of the anterior pituitary hormones may be attributable to organic defects or have an idiopathic etiology and may occur as a single hormonal problem or in combination with other hormonal disorders. The clinical manifestations depend on the hormones involved and the age of onset. Panhypopituitarism is often defined clinically as the loss of all anterior pituitary hormones, leaving only posterior function intact (Toogood and Stewart, 2008). Children with panhypopituitarism should wear medical identification, such as a bracelet. Hypopituitarism is diminished or deficient secretion of pituitary hormones. The consequences of the condition depend on the degree of dysfunction and can lead to gonadotropin deficiency with absence or regression of secondary sex characteristics; growth hormone (GH) deficiency, in which children display retarded somatic growth; thyroid-stimulating hormone (TSH) deficiency, which produces hypothyroidism; and corticotropin deficiency, which results in manifestations of adrenal hypofunction. Hypopituitarism can result from any of the conditions listed in Box 29-1. The most common organic cause of pituitary undersecretion is tumors in the pituitary or hypothalamic region, especially the craniopharyngiomas. Congenital hypopituitarism can be seen in newborn infants, often as a result of birth trauma. Symptoms of hypoglycemia and seizure activity often manifest within the first 24 hours after birth (Toogood and Stewart, 2008). Idiopathic hypopituitarism, or idiopathic pituitary growth failure, is usually related to GH deficiency, which inhibits somatic growth in all cells of the body (Miller and Zimmerman, 2004). Growth failure is defined as an absolute height of less than −2 standard deviation (SD) for age or a linear growth velocity consistently less than −1 SD for age. When this occurs without the presence of hypothyroidism, systemic disease, or malnutrition, then an abnormality of the GH–insulin-like growth factor (IGF-I) axis should be considered (Richmond and Rogol, 2008). Not all children with short stature have GH deficiency. In most instances, the cause is either familial short stature or constitutional growth delay. Familial short stature refers to otherwise healthy children who have ancestors with adult height in the lower percentiles. Constitutional growth delay refers to individuals (usually boys) with delayed linear growth, generally beginning as a toddler, and skeletal and sexual maturation that is behind that of age mates (Halac and Zimmerman, 2004; Miller and Zimmerman, 2004). Typically, these children will reach normal adult height. Often there is a history of a similar pattern of growth in one of the child’s parents or other family members. The untreated child will proceed through normal changes as expected on the basis of bone age. Although treatment with GH is not usually indicated, its use has become controversial, especially in relation to parental and child requests for treatment to accelerate growth. Children with hypopituitarism generally grow normally during the first year and then follow a slowed growth curve that is below the third percentile. Skeletal proportions and weight are normal for the age, but these children may appear younger than their chronologic age. Dentition is delayed, and teeth may be overcrowded and malpositioned because of the undeveloped jaw. Sexual development is usually delayed but is otherwise normal unless the gonadotropin hormones are deficient. Growth may extend into the third or fourth decade of life, but permanent height is usually diminished if the disorder is left untreated. Symptoms such as headache and vision changes may indicate the presence of a tumor. Clinical manifestations of panhypopituitarism are listed in Box 29-1. A complete diagnostic evaluation should include a family history, a history of the child’s growth patterns and previous health status, physical examination, psychosocial evaluation, radiographic surveys, and endocrine studies. Accurate measurement of height (using a calibrated stadiometer) and weight and comparison with standard growth charts are essential. Multiple height measures reflect a more accurate assessment of abnormal growth patterns (Box 29-2) (Hall, 2000). Parental height and familial patterns of growth are important clues to diagnosis. A skeletal survey in children younger than 3 years of age and radiographic examination of the hand–wrist for centers of ossification (bone age) (Box 29-3) in older children are important in evaluating growth. Definitive diagnosis is based on absent or subnormal reserves of pituitary GH. Because GH levels are variable in children, GH stimulation testing is usually required for diagnosis. Initial assessment of the serum IGF-I and IGF binding protein 3 (IGFBP3) indicates a need for further evaluation of GH dysfunction if levels are less than −1 SD below the mean for age. It is recommended that GH stimulation tests be reserved for children with low serum IGF-I and IGFBP3 levels and poor growth who do not have other causes for short stature (Richmond and Rogol, 2008). GH stimulation testing involves the use of pharmacologic agents such as levodopa, clonidine, arginine, insulin, propranolol, or glucagon to provoke the release of GH (Kliegman, Stanton, St. Geme, and others, 2011). Children with poor linear growth, delayed bone age, and abnormal GH stimulation tests are considered GH deficient. Treatment of GH deficiency caused by organic lesions is directed toward correction of the underlying disease process (e.g., surgical removal or irradiation of a tumor). The definitive treatment of GH deficiency is replacement of GH, which is successful in 80% of affected children. Biosynthetic GH is administered subcutaneously on a daily basis. Growth velocity increases in the first year and then declines in subsequent years. Final height is likely to remain less than normal (Bryant, Baxter, Cave, and others, 2007), and early diagnosis and intervention are essential (Leschek, Rose, Yanovski, and others, 2004). The decision to stop GH therapy is made jointly by the child, family, and health care team. Growth rates of less than 1 inch per year and a bone age of more than 14 years in girls and more than 16 years in boys are often used as criteria to stop GH therapy (Kliegman, Stanton, St. Geme, and others, 2011). Children with other hormone deficiencies require replacement therapy to correct the specific disorders. Children undergoing hormone replacement require additional support. The nurse should provide education for patient self-management during the school-age years. Nursing functions include family education concerning medication preparation and storage, injection sites, injection technique, and syringe disposal (see Chapter 22). Administration of GH is facilitated by family routines that include a specific time of day for the injection. Younger children may enjoy using a calendar and colorful stickers to designate received injections. Professionals and families can find resources for research, education, support, and advocacy from the Human Growth Foundation.* The treatment is expensive, but the cost is often partially covered by insurance if the child has a documented deficiency. Children with panhypopituitarism should be advised to wear medical identification at all times. If a lesion is present, surgery is performed to remove the tumor when feasible. Other therapies aimed at destroying pituitary tissue include external irradiation and radioactive implants. New pharmacologic agents have evolved and may be use in combination with other therapies (Natchtigall, Delgado, Swearingen, and others, 2008). Depending on the extent of surgical extirpation and degree of pituitary insufficiency, hormone replacement with thyroid extract, cortisone, and sex hormones may be necessary. Manifestations of sexual development before age 9 years in boys or age 8 years in girls have traditionally been considered precocious development, and these children were recommended for further evaluation (Kempers and Otten, 2002; Midyett, Moore, and Jacobson, 2003). Recent examination of the age limit for defining when puberty is precocious reveals that the onset of puberty in girls is occurring earlier than previous studies have documented (Biro, Huang, Crawford, and others, 2006; Slyper, 2006). The mean onset of puberty was 10.2 years in white girls and 9.6 years in African-American girls. Based on these findings, precocious puberty evaluation for a pathologic cause should be performed for white girls younger than 7 years of age or for African-American girls younger than 6 years of age. No change in the guidelines for evaluation of precocious puberty in boys is recommended. However, recent data suggest that boys may be beginning maturation earlier as well (Herman-Giddens, 2006; Slyper, 2006). Normally, the hypothalamic-releasing factors stimulate secretion of the gonadotropic hormones from the anterior pituitary at the time of puberty. In boys, interstitial cell–stimulating hormone stimulates Leydig cells of the testes to secrete testosterone; in girls, follicle-stimulating hormone (FSH) and luteinizing hormone stimulate the ovarian follicles to secrete estrogens (Nebesio and Eugster, 2007). This sequence of events is known as the hypothalamic–pituitary–gonadal axis. If for some reason the cycle undergoes premature activation, the child will display evidence of advanced or precocious puberty. Causes of precocious puberty are found in Box 29-4.
The Child with Endocrine Dysfunction

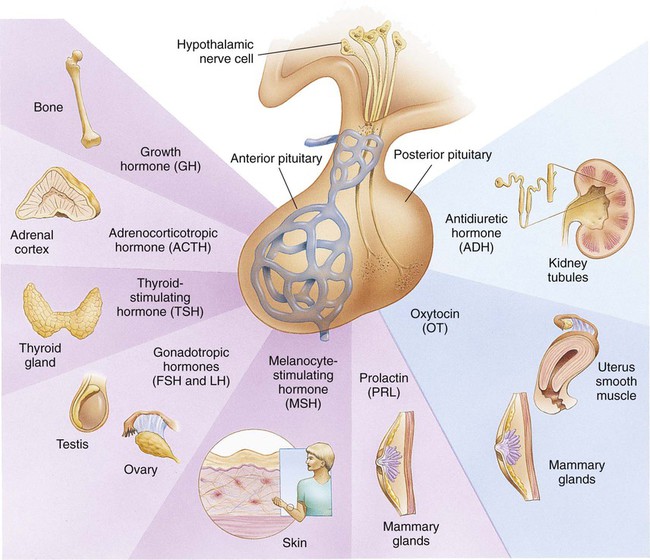
The Endocrine System
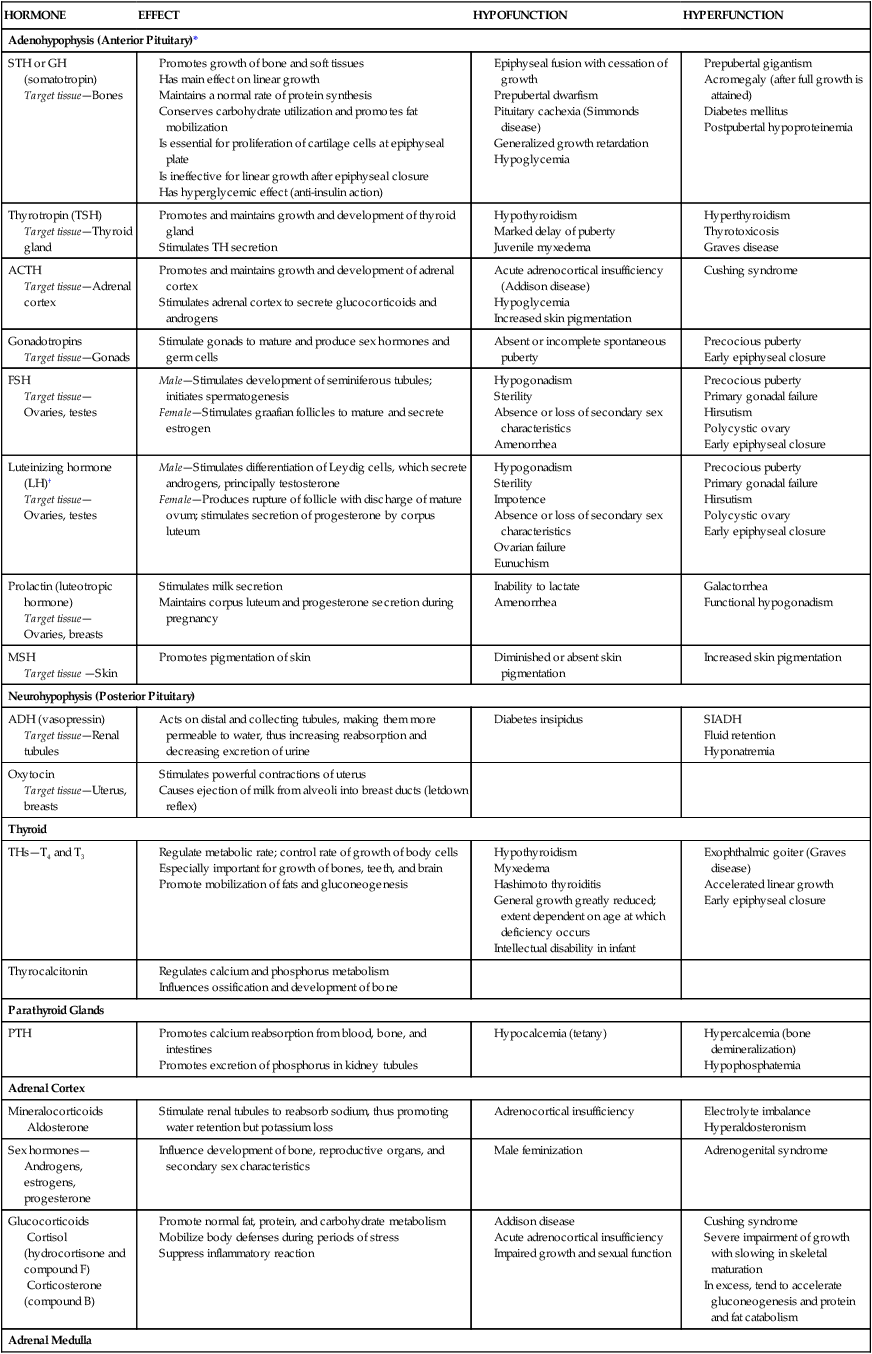
HORMONE
EFFECT
HYPOFUNCTION
HYPERFUNCTION
Adenohypophysis (Anterior Pituitary)*
STH or GH (somatotropin)
Target tissue—Bones
Thyrotropin (TSH)
Target tissue—Thyroid gland
ACTH
Target tissue—Adrenal cortex
Gonadotropins
Target tissue—Gonads
FSH
Target tissue—Ovaries, testes
Luteinizing hormone (LH)†
Target tissue—Ovaries, testes
Prolactin (luteotropic hormone)
Target tissue—Ovaries, breasts
MSH
Target tissue —Skin
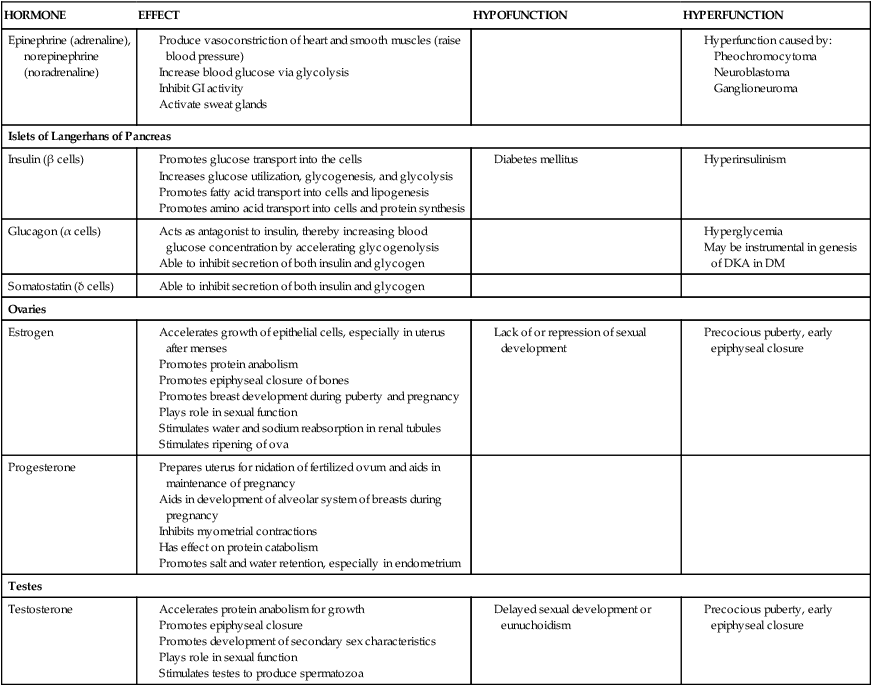
Neurohypophysis (Posterior Pituitary)
ADH (vasopressin)
Target tissue—Renal tubules
Oxytocin
Target tissue—Uterus, breasts
Thyroid
THs—T4 and T3
Thyrocalcitonin
Parathyroid Glands
PTH
Adrenal Cortex
Mineralocorticoids
Aldosterone
Sex hormones—Androgens, estrogens, progesterone
Glucocorticoids
Cortisol (hydrocortisone and compound F)
Corticosterone (compound B)
Adrenal Medulla
Epinephrine (adrenaline), norepinephrine (noradrenaline)
Islets of Langerhans of Pancreas
Insulin (β cells)
Glucagon (α cells)
Somatostatin (δ cells)
Ovaries
Estrogen
Progesterone
Testes
Testosterone
Hormones
Disorders of Pituitary Function
 Nursing Alert
Nursing Alert
Hypopituitarism
Clinical Manifestations
Diagnostic Evaluation
Therapeutic Management
Nursing Care Management
Child and Family Support
Pituitary Hyperfunction
Therapeutic Management
Precocious Puberty

The Child with Endocrine Dysfunction



Get Clinical Tree app for offline access