The Child with an Endocrine or Metabolic Alteration
Learning Objectives
After studying this chapter, you should be able to:
• List the major hormones of the endocrine system.
• Discuss nursing strategies to improve adherence with medication administration.
• Describe the signs and symptoms of hypothyroidism versus hyperthyroidism.
• Describe the psychosocial issues concerning children with precocious puberty.
• Compare and contrast type 1 diabetes mellitus and type 2 diabetes mellitus.

http://evolve.elsevier.com/McKinney/mat-ch
Clinical Reference
Review of the Endocrine System
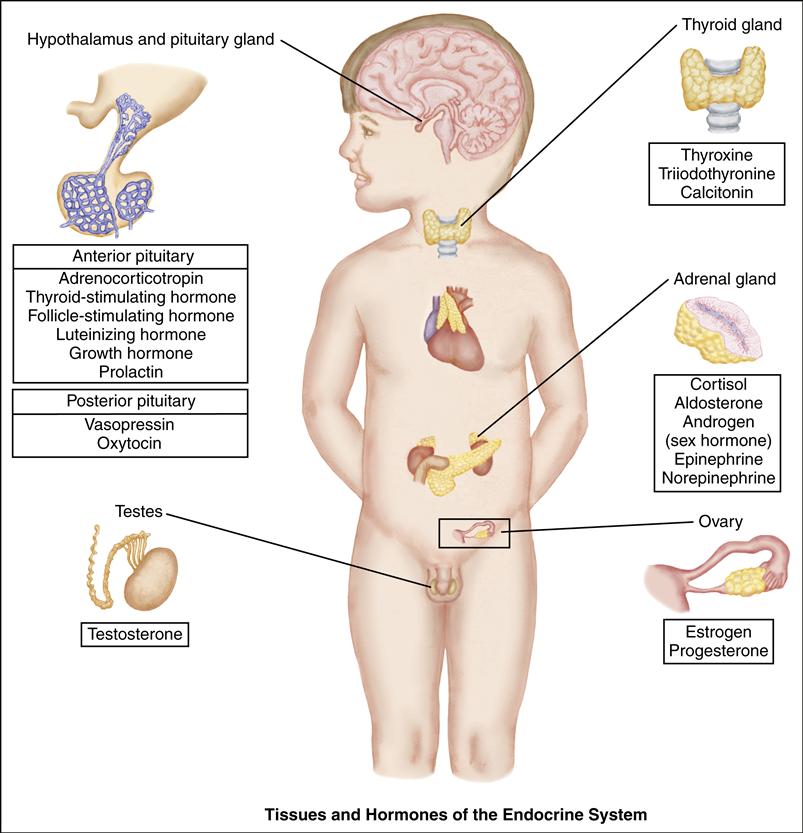
The endocrine system is composed of various tissues that produce and secrete chemicals called hormones. The hormones stimulate and regulate the actions of other tissues—the target tissues.
The endocrine system and the autonomic nervous system function in tandem to regulate growth, metabolism, and reproduction. The hypothalamic-pituitary axis controls their activities. The autonomic nervous system reacts to a stimulus, transmitting its message to the hypothalamus. In turn, the hypothalamus manufactures and secretes the appropriate hormonal factors. These are transmitted to the anterior pituitary gland, which then stimulates or inhibits the release of the involved hormones.
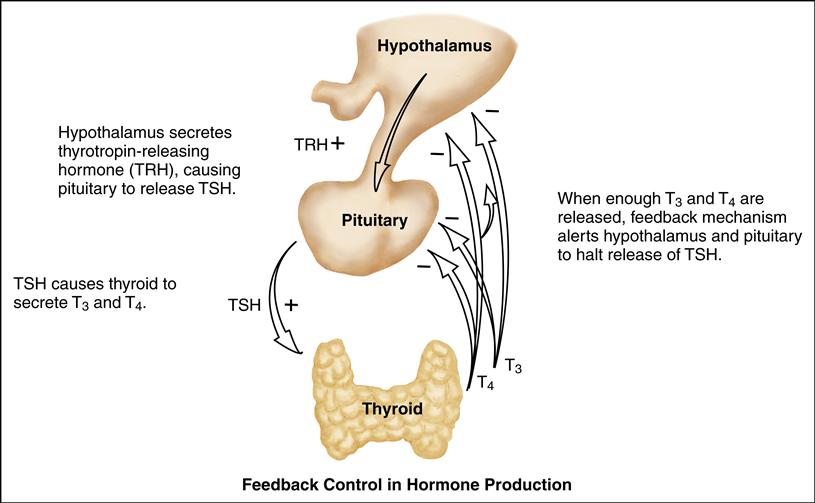
The principle of feedback control is involved in hormone production and secretion. In negative feedback, increasing levels of a specific hormone begin to inhibit the system responsible for releasing that hormone. As the hormonal secretion rises, the secretion and production of its stimulating hormone decrease. Conversely, when too little circulating hormone is present, the target gland is stimulated to secrete additional hormone.
The pituitary gland is composed of an anterior lobe and a posterior lobe. The anterior lobe secretes adrenocorticotropic hormone (ACTH), thyroid-stimulating hormone (TSH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), growth hormone (GH), and prolactin. Four of these hormones (ACTH, TSH, LH, and FSH) in turn stimulate their target glands to secrete the appropriate specific hormones. The posterior pituitary lobe stores and releases antidiuretic hormone (ADH) and oxytocin, which are synthesized by the hypothalamus.
Congenital malformations, infections, and neoplastic or autoimmune processes may disrupt normal endocrine function of the hypothalamus, pituitary gland, or target gland.
Fetal endocrine systems develop and function in utero. Portions of the endocrine system may be immature at birth but transition to more mature function after delivery.
Diagnostic Tests and Procedures
Diagnosing endocrine dysfunction usually involves laboratory testing. Serum hormone levels are measured to determine if the amounts are adequate, deficient, or excessive. Laboratory screening is useful for diagnosing disease and monitoring children on hormone therapy.
Normal hormone levels are related to the child’s age and stage of puberty. Because hormones are secreted at various times during the day or on a circadian rhythm, random blood samples may be difficult to interpret. Stimulation testing frequently demonstrates more accurate and definitive test results. With stimulation testing, a releasing factor or other agent is given to trigger the release or inhibition of a specific hormone. Serial blood sampling identifies the peak or trough level of the hormone, aiding in more accurate interpretation. A list of the common stimulation studies is provided in the accompanying table.
Other diagnostic tests include radiography and imaging techniques. Bone age radiographs can determine bone maturation, and from this, growth potential can be determined. Computed tomography (CT) scans and magnetic resonance imaging (MRI) are used to determine the presence of tumors or congenital malformations affecting the hypothalamus, pituitary, or target glands.
Accurate measurements of height and weight are essential when assessing the child for endocrine function. Evaluation of sexual development according to Tanner stages is also a part of the diagnostic workup (see Chapter 9). Developmental milestones and school performance should also be monitored because delays may be associated with endocrine disorders.
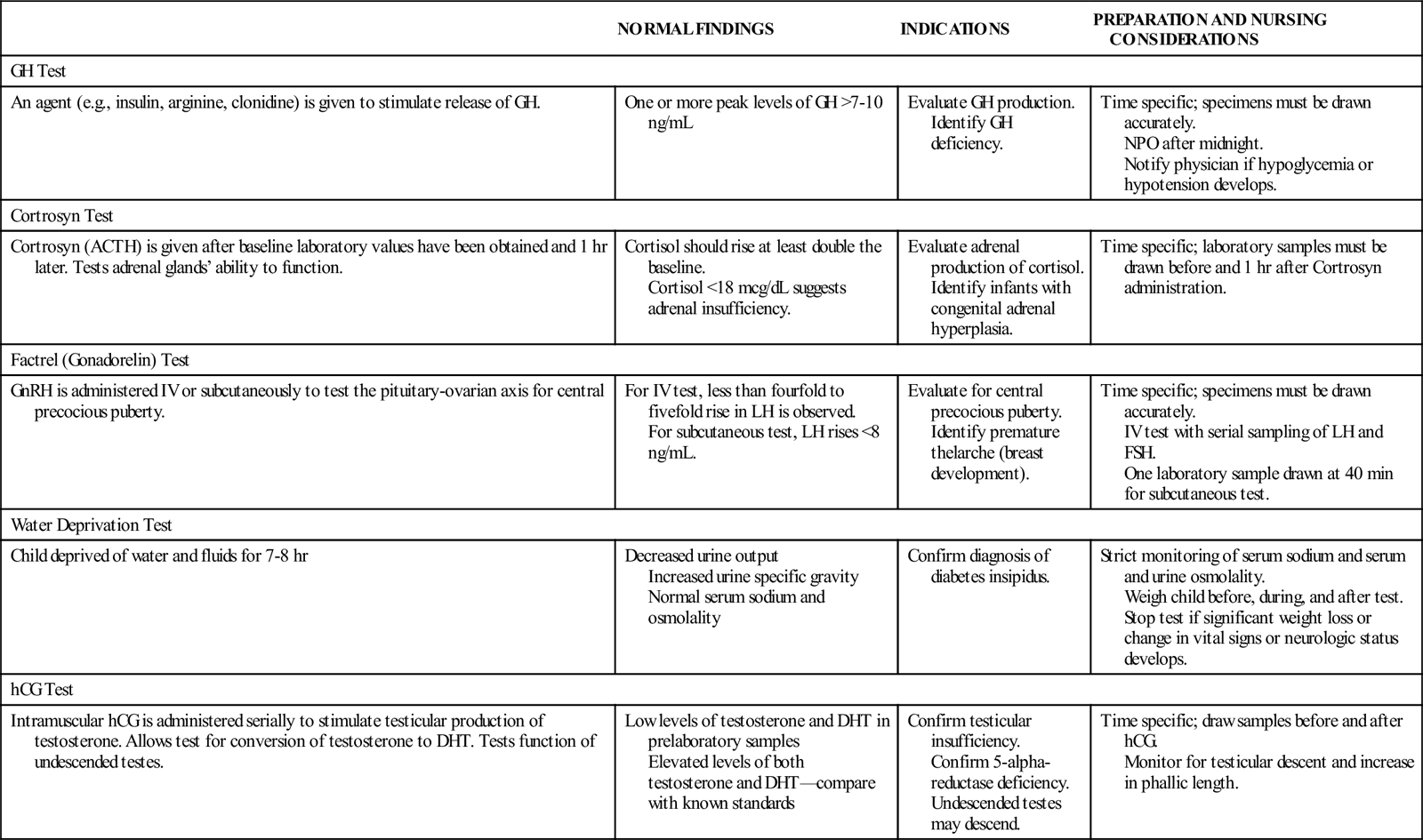
COMMON LABORATORY AND DIAGNOSTIC TESTS OF ENDOCRINE FUNCTION
| NORMAL FINDINGS | INDICATIONS | PREPARATION AND NURSING CONSIDERATIONS | |
| GH Test | |||
| An agent (e.g., insulin, arginine, clonidine) is given to stimulate release of GH. | One or more peak levels of GH >7-10 ng/mL | Evaluate GH production. Identify GH deficiency. | Time specific; specimens must be drawn accurately. NPO after midnight. Notify physician if hypoglycemia or hypotension develops. |
| Cortrosyn Test | |||
| Cortrosyn (ACTH) is given after baseline laboratory values have been obtained and 1 hr later. Tests adrenal glands’ ability to function. | Cortisol should rise at least double the baseline. Cortisol <18 mcg/dL suggests adrenal insufficiency. | Evaluate adrenal production of cortisol. Identify infants with congenital adrenal hyperplasia. | Time specific; laboratory samples must be drawn before and 1 hr after Cortrosyn administration. |
| Factrel (Gonadorelin) Test | |||
| GnRH is administered IV or subcutaneously to test the pituitary-ovarian axis for central precocious puberty. | For IV test, less than fourfold to fivefold rise in LH is observed. For subcutaneous test, LH rises <8 ng/mL. | Evaluate for central precocious puberty. Identify premature thelarche (breast development). | Time specific; specimens must be drawn accurately. IV test with serial sampling of LH and FSH. One laboratory sample drawn at 40 min for subcutaneous test. |
| Water Deprivation Test | |||
| Child deprived of water and fluids for 7-8 hr | Decreased urine output Increased urine specific gravity Normal serum sodium and osmolality | Confirm diagnosis of diabetes insipidus. | Strict monitoring of serum sodium and serum and urine osmolality. Weigh child before, during, and after test. Stop test if significant weight loss or change in vital signs or neurologic status develops. |
| hCG Test | |||
| Intramuscular hCG is administered serially to stimulate testicular production of testosterone. Allows test for conversion of testosterone to DHT. Tests function of undescended testes. | Low levels of testosterone and DHT in prelaboratory samples Elevated levels of both testosterone and DHT—compare with known standards | Confirm testicular insufficiency. Confirm 5-alpha-reductase deficiency. Undescended testes may descend. | Time specific; draw samples before and after hCG. Monitor for testicular descent and increase in phallic length. |
Pediatric endocrine disorders are generally managed in the outpatient setting. Most endocrine disorders are chronic conditions requiring long-term nursing management. The nurse assumes a role of both educator and advocate for the child. Also important for the care of children with chronic medical problems is a careful psychosocial evaluation on a regular basis.
Phenylketonuria
Phenylketonuria (PKU) is a genetic metabolic disorder that results in central nervous system (CNS) damage from toxic levels of phenylalanine in the blood. PKU is characterized by a deficiency of phenylalanine hydroxylase, the enzyme needed to convert phenylalanine to tyrosine.
Etiology
PKU is an autosomal recessive disorder and is manifested only in the homozygote (individual who inherited two identical genes for a specific trait). With both parents carrying the recessive gene, each pregnancy has a 25% chance that the child will have PKU.
Incidence
PKU occurs in approximately 1 in 15,000 births in the United States (Arnold, 2009). It is more prevalent in some European countries.
Manifestations
The underlying metabolic alterations begin to have an immediate effect on the infant, although signs may not be apparent until the infant is approximately 3 months old. The first sign may be digestive problems with vomiting. These infants also may have a musty or mousy odor to the urine, infantile eczema, hypertonia, and hyperactive behavior. Older children may have hypopigmentation of the hair, skin, and irises; they are commonly blond with light blue eyes. Intellectual impairment is a long-term consequence of untreated PKU.
Diagnostic Evaluation
Routine neonatal screening for PKU is mandatory in all 50 states of the United States. With early postpartum discharge, screening is often performed on infants younger than 2 days of age because of the concern that the infant will be lost to follow-up. Because the test depends on the accumulation of phenylalanine, screening done before the third day of life has a higher risk of a false-negative outcome. For this reason, testing should be done after the infant is 48 hours old, or, if done earlier, the test should be repeated at several days of age. Small quantities of blood are collected on filter paper cards. Screening is done by bacterial inhibition (Guthrie test) or chromatographic or fluorometric assays. A positive result is not diagnostic but indicates which infants should be evaluated further. PKU is characterized by serum phenylalanine levels greater than 20 mg/dL (normal level 2 mg/dL) (Arnold, 2009).
Therapeutic Management
Treatment should be instituted as soon as the diagnosis is confirmed because the best results are obtained with early treatment. Infants and children with PKU are treated with a special diet that restricts phenylalanine intake. Phenylalanine tolerance varies according to the infant and the severity of the enzyme deficiency. The goal of therapy is to keep the serum phenylalanine level at 2 to 6 mg/dL in infants and young children and 2 to 15 mg/dL in children older than 12 years of age. Phenylalanine intake should be limited while providing enough of this essential amino acid to meet the baby’s growth requirement. Dietary management must be started early in neonatal life because the untreated infant will show evidence of CNS damage by several weeks of age. The age at which the diet may be discontinued is a point of controversy. Most health care facilities in the United States recommend lifelong continuation of the diet (Arnold, 2009). Cofactor tetrahydrobiopterin (sapropterin [Kuvan]), a medication that can lower blood phenylalanine level, may be appropriate for some individuals with PKU (Arnold, 2009).
Another consideration is genetic counseling for women with PKU who become pregnant. Adolescent girls and women of childbearing age require counseling about fetal risks including mental deficiency, microcephaly, retarded growth, seizures, and an increased incidence of structural defects. The goal is to control phenylalanine levels before conception and maintain strict control during the pregnancy.
Nursing Considerations
Although a family history of PKU would alert the caregiver to an infant at risk, most infants with PKU are not identified at birth. Neonatal symptoms are usually not present. A screening test, part of the newborn screen done in all states, is the first diagnostic procedure. Newborn screenings usually include testing for PKU and congenital hypothyroidism, as well as any other screening tests mandated by local and state public health departments, such as sickle cell trait, galactosemia, and maple syrup urine disease. A positive screening result requires further diagnostic evaluation to verify the diagnosis.
A low-phenylalanine diet is begun immediately; the infant with PKU is fed a low-phenylalanine formula and, as foods are introduced, the child must follow a protein-restricted diet. The child must avoid high-protein foods such as meats, fish, eggs, cheese, milk, and legumes. Because protein is also present in grains, low-protein breads, cereals, and pastas are used. Dietary staples are vegetables, fruits, and starches. To avoid the consequences of insufficient protein for growth, children with PKU may take a phenylalanine-free protein supplement. The growth pattern and neurobehavior of the affected child must be monitored.
Follow-up is provided for all infants if the initial screening result is abnormal. The nurse assists with referral to a genetic center that is capable of diagnosing and treating the infant.
Phenylalanine requirements change rapidly in the first months of life. Parents are encouraged to adhere to monitoring requirements for the infant diagnosed with PKU. Rigid regimens for diet control will not be successful unless the family accepts the changes required. The nurse helps the family deal with lifestyle changes by initiating referrals as needed (e.g., to social service agencies, registered dietitian, support groups). On-line support groups and chat rooms provide ideas for adapting recipes; specialized cookbooks are also available.
The nurse encourages the parents to express their feelings about the infant’s diagnosis and the risk of PKU in future children. Family members need support to recognize the problems caused by the disease and identify strategies for dealing with the stress of having a child with a chronic illness. Physical measurements and neurologic and intellectual development should be documented through standardized testing. If control of the phenylalanine level is established early, normal infant growth and development should occur.
Inborn Errors of Metabolism
In addition to PKU, there are other genetically transmitted metabolic diseases that rarely occur in newborns (Table 51-1). Nurses should create a climate in which parents can express their feelings about the lifelong care of their child, as well as concerns for future pregnancies. Families with affected infants are referred to genetic counseling centers. Many of these infants are identified through universal newborn screening or screening specific for at-risk infants. Additional nursing care is related to the particular disorder but is similar to that for the child with PKU.
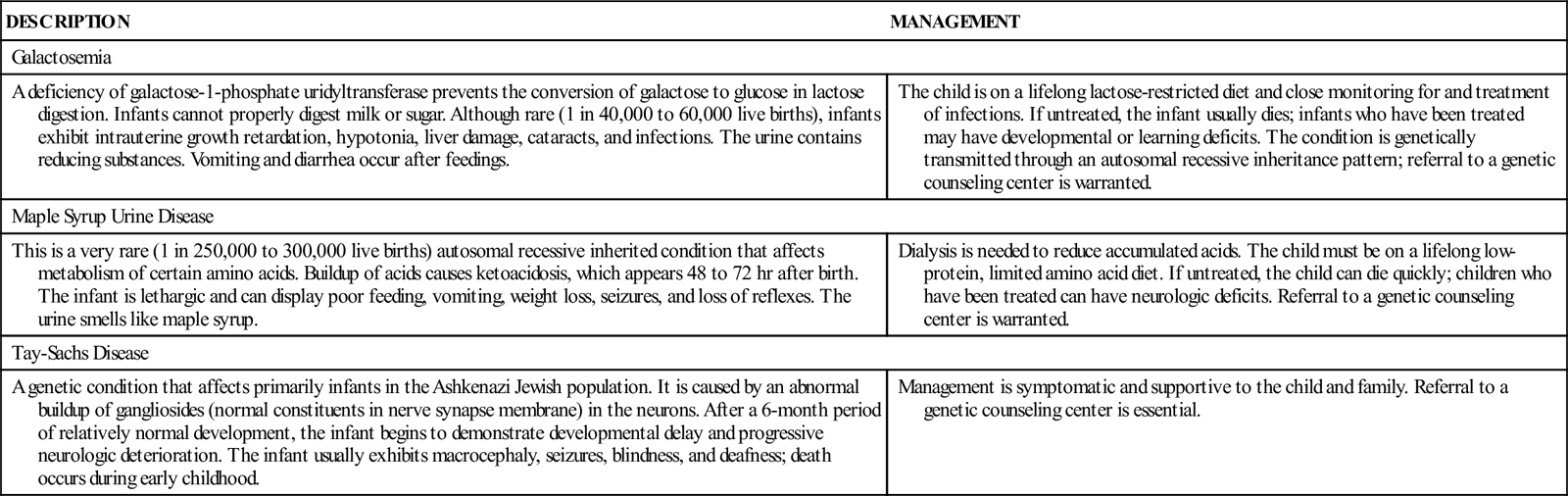
TABLE 51-1
| DESCRIPTION | MANAGEMENT |
| Galactosemia | |
| A deficiency of galactose-1-phosphate uridyltransferase prevents the conversion of galactose to glucose in lactose digestion. Infants cannot properly digest milk or sugar. Although rare (1 in 40,000 to 60,000 live births), infants exhibit intrauterine growth retardation, hypotonia, liver damage, cataracts, and infections. The urine contains reducing substances. Vomiting and diarrhea occur after feedings. | The child is on a lifelong lactose-restricted diet and close monitoring for and treatment of infections. If untreated, the infant usually dies; infants who have been treated may have developmental or learning deficits. The condition is genetically transmitted through an autosomal recessive inheritance pattern; referral to a genetic counseling center is warranted. |
| Maple Syrup Urine Disease | |
| This is a very rare (1 in 250,000 to 300,000 live births) autosomal recessive inherited condition that affects metabolism of certain amino acids. Buildup of acids causes ketoacidosis, which appears 48 to 72 hr after birth. The infant is lethargic and can display poor feeding, vomiting, weight loss, seizures, and loss of reflexes. The urine smells like maple syrup. | Dialysis is needed to reduce accumulated acids. The child must be on a lifelong low-protein, limited amino acid diet. If untreated, the child can die quickly; children who have been treated can have neurologic deficits. Referral to a genetic counseling center is warranted. |
| Tay-Sachs Disease | |
| A genetic condition that affects primarily infants in the Ashkenazi Jewish population. It is caused by an abnormal buildup of gangliosides (normal constituents in nerve synapse membrane) in the neurons. After a 6-month period of relatively normal development, the infant begins to demonstrate developmental delay and progressive neurologic deterioration. The infant usually exhibits macrocephaly, seizures, blindness, and deafness; death occurs during early childhood. | Management is symptomatic and supportive to the child and family. Referral to a genetic counseling center is essential. |
Data from Rezvani, I., & Rezvani, G. (2011). An approach to inborn errors of metabolism. In R. Kliegman, B. Stanton, J. St. Geme, et al., (Eds.), Nelson textbook of pediatrics (19th ed., pp. 416-418). Philadelphia: Saunders.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia (CAH) is a group of disorders in which the adrenal gland is not able to manufacture adequate glucocorticoid and, while working to make glucocorticoid, produces excess androgens. CAH is caused by a defect in the enzymatic pathway of adrenal steroid production. Diminished glucocorticoid production prompts increased ACTH production, further increasing adrenal androgen excess. Mineralocorticoid production may be normal or low.
Etiology
Infants with diminished mineralocorticoid production will waste salt through the kidneys, resulting in a “salt-wasting” crisis. This occurs with the more life-threatening form of CAH, which makes up 50% of cases and is the most common type (Wilson, 2010). Salt-wasting crisis results in hypovolemia, low serum sodium levels, hypotensive crisis, and hyperkalemia. Several enzymatic defects have been identified, the most common being 21-hydroxylase deficiency (cytochrome P21 [CYP21]). CAH is an autosomal recessive condition.
Manifestations
CAH is marked by ambiguous genitalia of the newborn female infant; postnatal virilization in both sexes; and salt-wasting crisis (in the first few weeks of life) with low serum sodium, high serum potassium, hypovolemia, and hypotensive crisis. Simple virilizing CAH is not associated with a salt-wasting crisis and manifests with a muscular body, advanced bone age, and premature pubic hair. Typically this form is seen later in infancy or early childhood. Untreated or poorly treated CAH can result in an advanced bone age with ultimate adult short stature. A milder form of CAH, 3-beta-hydroxysteroid dehydrogenase (3β-HSD), may become symptomatic during childhood or adolescence, with the child manifesting hirsutism, menstrual irregularities, or delayed menses.
Diagnostic Evaluation
The finding of ambiguous genitalia in the newborn infant should raise the possibility of CAH. The diagnosis is confirmed by elevated values of 17-hydroxyprogesterone, a glucocorticoid precursor. CAH is a part of newborn screening in many states. An appropriate evaluation includes obtaining serum electrolyte, carbon dioxide, and renin levels, and performing a physical examination. Serum sodium levels in the infant suspected of CAH will be low, with elevated serum potassium. Serum renin levels will be elevated, indicating mineralocorticoid deficiency. A karyotype to determine genetic sex may be indicated depending on the degree of genital ambiguity.
Therapeutic Management
An accurate diagnosis and prompt treatment of fluid and electrolyte abnormalities may avert a salt-wasting crisis. The child with CAH requires lifelong glucocorticoid therapy. An oral glucocorticoid (hydrocortisone acetate, cortisone acetate) dosage is prescribed on the basis of body size and is given two or three times per day in either a liquid suspension or tablet form. For children with salt-wasting CAH, mineralocorticoid replacement therapy is required using fludrocortisone acetate (Florinef) which is taken once or twice daily. Therapy effectiveness is evaluated with serum electrolyte, 17-hydroxyprogesterone level, and renin levels. Special sick-day instructions should be provided to the family. The glucocorticoid dosage is usually doubled or tripled when the child is ill, has a broken bone, or is undergoing a surgical procedure. Bone age radiographs are performed yearly to assess skeletal maturity; poor adherence to the medication regimen or undertreatment can result in advanced bone age and decreased final adult height.
Nursing Considerations
All newborn girls should be assessed for ambiguous genitalia; fused labia, enlarged clitoris, or migration of urethral opening. Infant boys with unexplained dehydration and low serum sodium levels should be considered to have adrenal insufficiency, and undergo careful assessment of fluid and electrolyte status.
 SAFETY ALERT
SAFETY ALERT
Congenital Adrenal Hyperplasia
Infant girls with ambiguous genitalia might require reconstructive surgery. Depending on degree of virilization, surgical correction may be recommended in infancy or in early puberty. When appropriate, the nurse reassures the parents that the infant has appropriate internal structures and that external structures can be corrected surgically. Parents are encouraged to express their concerns. The nurse helps to facilitate parent-infant attachment.
Older children receiving glucocorticoid replacement therapy are assessed for linear growth and signs of early puberty. Serial height measurements can provide data about the adequacy of glucocorticoid supplementation. In children with salt-wasting CAH, serum renin levels should be closely monitored; effective treatment with mineralocorticoids will maintain these levels in or near the normal range (White, 2011). Nonadherence can cause early virilization, increased growth velocity, diminished final adult height, and menstrual irregularities in girls. Blood pressure monitoring is important for children receiving mineralocorticoid replacement therapy.
The nurse carefully instructs the parents about replacement hormone administration and the timing of medication. Parents are included in the development of a plan for sick-day dosages of medications. The infant with salt-wasting CAH may require salt supplements; the family needs instruction on preparation of these supplements.
Follow-up evaluations with the endocrinologist are scheduled every 2 to 3 months in infancy and every 4 to 6 months in the older child. Parents of the child with CAH should be referred to a genetic counselor if they plan more pregnancies because future children are at risk for CAH. In utero treatment is available to prevent virilization of the female fetus. If effective, this prenatal treatment eliminates the need for surgical correction of ambiguous genitalia in the affected female infant.
Adolescents are encouraged to assume increasing responsibility for medication administration. The nurse strongly emphasizes the importance of compliance. Surgical reconstruction of the genitalia and vaginal dilation may be required in the adolescent years. Careful explanations and preparation for these procedures will reassure affected adolescents and help them understand the expected outcomes following the procedures.
Congenital Hypothyroidism
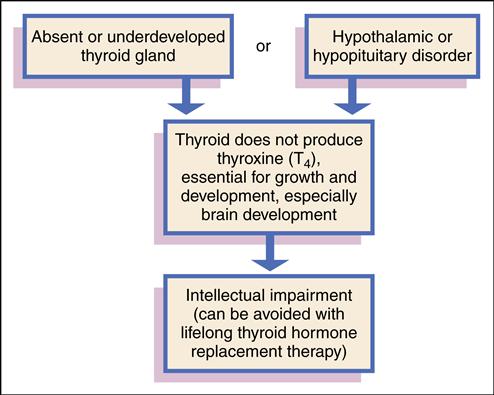
Congenital hypothyroidism is a condition in which the thyroid gland does not produce sufficient thyroid hormone to meet the body’s metabolic needs. The condition is present from birth and, if not treated, can lead to intellectual impairment.
Etiology
Congenital hypothyroidism is caused by an absent (aplastic), underdeveloped, or ectopic thyroid gland. This group of congenital defects is referred to as thyroid dysgenesis. For unknown reasons, the fetal thyroid gland fails to develop properly or fails to migrate to the appropriate location. Other rare causes are hypothalamic or pituitary disorders in which TSH secretion is insufficient to stimulate the thyroid gland. Biochemical defects in thyroid hormone production also cause congenital hypothyroidism. Maternal intake of medications, such as propylthiouracil (PTU), during pregnancy to control maternal hyperthyroidism can cause transient hypothyroidism in the infant. Transfer of maternal antibodies to the fetus may also cause transient hypothyroidism (Postellon, 2010).
Incidence
The incidence of congenital hypothyroidism in the United States is approximately 1 in 4000 live births (Postellon, 2010). Because untreated hypothyroidism causes intellectual impairment, all states have mandatory newborn screening programs to diagnose hypothyroidism before symptoms occur. Early detection and treatment favor increased intellectual function. Most occurrences are spontaneous, with a smaller percentage having a genetic (autosomal recessive) inheritance that results in defective thyroxine synthesis (Postellon, 2010).
Manifestations
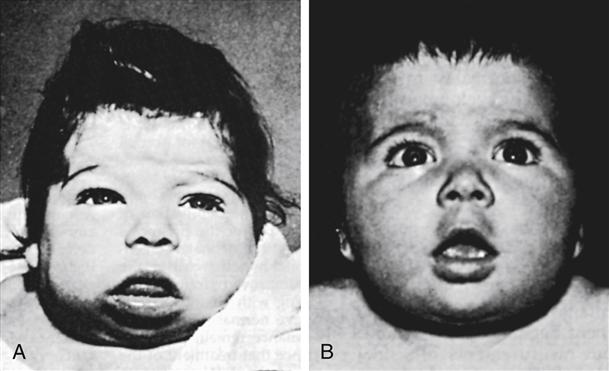
The infant with congenital hypothyroidism often displays signs of skin mottling, a large fontanel, a large tongue, hypotonia, slow reflexes, and a distended abdomen (Figure 51-1). Other signs and symptoms include prolonged jaundice, lethargy, constipation, feeding problems, coldness to touch, umbilical hernia, hoarse cry, and excessive sleeping. The infant with congenital hypothyroidism may have none of these signs or symptoms; the newborn screening test is essential to recognize these infants.
Diagnostic Evaluation
Congenital hypothyroidism is usually diagnosed by testing of the serum thyroxine (T4) level with newborn screening. Ideally, testing should be performed at 2 to 6 days of age. Tests performed sooner than 48 hours after delivery may be falsely interpreted because of the rise in TSH immediately after birth as part of the normal newborn transition. For a newborn with a low T4 value, a TSH level will be obtained. A low T4 level with TSH elevation is indicative of congenital hypothyroidism; further testing is often done to determine the cause (LaFranchi, 2011b). Thyroid scans can identify any functioning thyroid tissue. Treatment should never be delayed while waiting for scan results.
Therapeutic Management
Treatment of children with congenital hypothyroidism consists of lifelong thyroid hormone replacement, usually in the form of levothyroxine. It is given as a single daily oral dose that varies with the weight and age of the child or adult (Postellon, 2010). The dosage is titrated to maintain TSH and T4 in a normal range.
Nursing Care
The Infant with Congenital Hypothyroidism
Assessment
Nursing care of the infant with congenital hypothyroidism involves assessing growth and development and ensuring adherence to the prescribed medication regimen. Nurses can play a major role in recognizing the infant with hypothyroidism. Intellectual impairment caused by untreated hypothyroidism cannot be reversed, but it can be prevented through early identification and proper treatment. In general, infants with hypothyroidism are evaluated every 1 to 2 months for the first year of life and then every 3 to 6 months thereafter. The nurse should obtain accurate measurements of height, weight, and head circumference at each visit. Frequent developmental assessments are also essential.
Nursing Diagnosis and Planning
The following nursing diagnoses and expected outcomes may be appropriate for the infant with congenital hypothyroidism and the infant’s parents:
Expected Outcomes
The parents will demonstrate the ability to monitor their infant for signs and symptoms of hypothyroidism and hyperthyroidism and give thyroid medication properly. The parents will verbalize an understanding of normal growth and development milestones and their child’s lifelong needs for care and follow-up.
Expected Outcomes
As a result of appropriate disease management, the infant will demonstrate growth and developmental milestones appropriate for age.
Expected Outcome
As a result of disease management, the infant will maintain a temperature within normal range.
Interventions
The nurse instructs family members on the importance of medication adherence emphasizing that the medication is necessary for the child’s growth, especially for the rapidly developing brain.
The family is taught how to administer levothyroxine orally as a single daily dose. It can be dissolved in a small amount of water and given by syringe or placed into the nipple of a baby bottle (see Chapter 38). When the infant is older, the medication can be given in a spoonful of baby food. Toddlers can usually chew tablets without difficulty. If the infant or child vomits within 1 hour of taking medication, the dose should be readministered. Frequently missed doses can lead to developmental delays and poor growth.
 NURSING QUALITY ALERT
NURSING QUALITY ALERTThe nurse also teaches the parents the signs and symptoms of both hypothyroidism and hyperthyroidism and when to notify the physician if symptoms occur. Hyperthyroidism can develop in infants receiving too much medication. Parents need to be taught to count their child’s pulse and notify their health care provider if the rate is greater than the recommended parameter.
Because hypothyroidism is a lifelong condition, school-age children and teenagers should be made aware of the importance of taking their medication and of keeping regular follow-up appointments with the physician.
Evaluation
Acquired Hypothyroidism
Hypothyroidism is a condition in which the thyroid gland produces an inadequate amount of thyroid hormone to meet the body’s metabolic needs.
Etiology
Hashimoto thyroiditis, a common cause of acquired hypothyroidism, is usually associated with a goiter. It is the result of an autoimmune process. Other causes of acquired hypothyroidism include surgical thyroidectomy, radioactive iodine therapy for hyperthyroidism, radiation therapy for malignancies, and excessive iodine ingestion. Less frequently, decreased TSH secretion by the pituitary gland or decreased thyrotropin-releasing hormone (TRH) secretion by the hypothalamus causes hypothyroidism.
Autoimmune thyroiditis is the most common cause of acquired hypothyroidism in children and adolescents (LaFranchi, 2011b). It often occurs in families with a history of thyroid disease. Other family members may have positive thyroid antibodies. Thyroiditis is more common in girls, with as many as 10% of young girls exhibiting signs of autoimmune thyroid dysfunction, most often chronic lymphocytic thyroiditis (Ferry & Bauer, 2010).
Pathophysiology
Circulating autoantibodies known as thyroid-blocking immunoglobulins decrease thyroid gland production of triiodothyronine (T3) and T4. These antibodies bind at the TSH receptor sites on the thyroid gland, resulting in decreased thyroid hormone production. The cause of antibody production is unknown.
In contrast to congenital hypothyroidism, adverse effects from hypothyroidism acquired after 2 to 3 years of age are often reversible. Goiter, an enlarged thyroid gland, occurs in response to increased TSH secretion, autoimmune attack of the thyroid gland, or goitrogens.
Manifestations
Clinical manifestations of hypothyroidism include goiter (one lobe frequently larger than the other); dry, thick skin; coarse, dull hair; fatigue; cold intolerance; constipation; weight gain; decreased linear growth; edema of face, eyes, and hands; and irregular or delayed menses (Box 51-1).
Diagnostic Evaluation
Elevated TSH and low T4 levels are diagnostic of hypothyroidism. Elevated TSH level is the most sensitive indicator of primary hypothyroidism.
Thyroiditis is diagnosed by the presence of circulating thyroid antibodies and is usually associated with a firm goiter. Initially TSH is elevated with normal T4 levels, although T4 decreases over time. With secondary or tertiary hypothyroidism, TSH is not elevated; therefore, thyroid-releasing hormone stimulation testing is usually required for diagnosis.
Therapeutic Management
Management of the child with hypothyroidism involves thyroid hormone replacement, usually with levothyroxine. Dosage varies according to the child’s age and weight and is given as a single daily dose. The dose is titrated to maintain T4 in the upper half of the normal range and to maintain TSH in the normal range for age.
Nursing Care
The Child with Acquired Hypothyroidism
Assessment
Care of the child with acquired hypothyroidism includes assessing response to treatment and adherence to the medication regimen. With treatment, the goiter should decrease in size. Signs and symptoms of hypothyroidism should also resolve with adequate thyroid hormone replacement. Monitoring height and weight, and conducting developmental screening at each clinic visit assesses the child’s growth and development. The nurse should monitor school performance as well and maintain contact with the school nurse.
Nursing Diagnosis and Planning
The following nursing diagnoses and expected outcomes may be appropriate for a child with acquired hypothyroidism:
Expected Outcome
The child will maintain regular bowel movements of normal consistency as basal metabolic rate improves.
Expected Outcome
The child will maintain normal energy levels for age, as evidenced by the ability to exercise at the same level as peers.
Expected Outcomes
The child will verbalize feelings about body changes and will accept reassurances that changes will resolve with treatment.
Expected Outcome
As a result of appropriate disease management, the child will maintain normal body temperature.
Interventions
Parents and children who are school age or older should be instructed on the correct dose and timing of thyroid medication. Thyroid hormone levels are usually checked every 3 to 6 months. Laboratory values within the normal range indicate good response to therapy. The nurse educates the older child and parents on the signs and symptoms of hypothyroidism and hyperthyroidism and to notify the physician if symptoms occur. The child is reassured that signs such as constipation, fatigue, and weight gain will resolve as the medication becomes effective.
Evaluation
Hyperthyroidism (Graves Disease)
Graves disease is an autoimmune condition in which excessive thyroid hormones are produced by an enlarged thyroid gland. It is the most common cause of hyperthyroidism in children.
Incidence
The incidence of Graves disease in children is approximately 1 in 5000, with girls being five times more likely than boys to acquire the condition. Peak age for acquiring the condition is between 10 and 14 years (Ferry & Levitsky, 2010). Graves disease may also have a familial tendency (Ferry & Levitsky, 2010). Children with autoimmune disease are at risk for other autoimmune disorders. Neonatal Graves disease is associated with maternal hyperthyroidism and is relatively uncommon.
Pathophysiology
Circulating autoantibodies known as thyroid-stimulating immunoglobulins (TSIs) stimulate the thyroid gland to make T3 and T4. These antibodies bind to the TSH receptor sites on the thyroid gland, resulting in excessive thyroid hormone production. The cause of antibody production is unknown. In newborns, maternal TSI is transferred through the placenta to the fetus. TSIs bind to the TSH receptor, causing neonatal hyperthyroidism.
Manifestations
Goiter, increased appetite, weight loss, nervousness, diarrhea, increased perspiration, heat intolerance, increased heart rate, muscle weakness, palpitations, tremors, exophthalmos, poor attention span, and behavior or school problems are common in Graves disease (see Box 51-1). In the neonate, irritability, tachycardia, hypertension, voracious appetite with poor weight gain, flushing, prominent eyes, and thyroid enlargement are major signs. These are self-limiting signs, but cardiac failure and death can occur if the signs are unrecognized or poorly treated.
 NURSING QUALITY ALERT
NURSING QUALITY ALERTDiagnostic Evaluation
Elevated serum T4 levels and suppressed TSH levels, associated with signs and symptoms of hyperthyroidism, suggest Graves disease. Autoantibodies to thyroid tissue usually are positive. Thyroid uptake of radioactive iodine is increased.
Therapeutic Management
The three approaches to the management of Graves disease are antithyroid drug therapy, radioactive iodine, or surgery. Antithyroid drug therapy with methimazole is the treatment of choice for childhood hyperthyroidism. PTU, because of its increased risk for liver toxicity, should only be used in children who are allergic to methimazole (Ferry & Levitsky, 2010). These drugs act by blocking thyroid hormone production by the thyroid gland (Ferry & Levitsky, 2010). The medications usually are given three times per day, and they lower thyroid hormone levels in several weeks. Minor adverse effects include arthralgia, skin rash, pruritus, and gastric intolerance. Major adverse effects may include neutropenia, hepatotoxicity, and hypothyroidism.
A second approach to management is oral radioactive iodine treatment. Radioactive iodine (131I) is given as an oral solution. It is typically used in children older than 10 years. With this therapy, the radioactive iodine is absorbed and concentrated by the thyroid gland, destroying the thyroid tissue in approximately 6 to 18 weeks. Hyperthyroid symptoms may intensify briefly after treatment. Hypothyroidism can result once the thyroid gland is irradiated, necessitating thyroid replacement therapy.
Subtotal or partial thyroidectomy, the surgical removal of thyroid gland tissue, is the third form of management. Lugol’s solution (potassium iodide), given 10 to 14 days before surgery, decreases the gland’s vascularity. Surgery carries the risk of injury to the parathyroid glands, resulting in hypocalcemia. Calcium levels are monitored after surgery.
Recurrence of hyperthyroidism is uncommon but possible. Affected children also have a 60% to 80% chance for developing hypothyroidism, which can be treated with thyroid replacement therapy.
Follow-up evaluations correlate with response to therapy. As thyroid functions normalize, follow-up endocrine evaluations are recommended once or twice per year.
Nursing Care
The Child with Hyperthyroidism
Assessment
The treatment goals consist of normalizing thyroid hormone levels, alleviating symptoms of hyperthyroidism, and decreasing the goiter. The nurse should assess for adherence to medical therapy and determine if the family understands that medical therapy might take several weeks to decrease thyroid hormone action. The child is closely monitored for adrenergic signs and symptoms. Propranolol, a beta-adrenergic blocker, may be prescribed to decrease adrenergic signs and symptoms (tachycardia, heat intolerance, tremor) until the antithyroid medication takes effect.
A child being treated with PTU has an increased risk of neutropenia and hepatotoxicity; regular blood counts and liver function studies are done to assess these risks. The nurse assesses the child for fever, joint pain, edema, rash, or excessive bruising. A child who acquires a fever or sore throat while receiving PTU should be evaluated by a physician and a complete blood count obtained.
Nursing Diagnosis and Planning
The following nursing diagnoses and expected outcomes may be appropriate for a child with hyperthyroidism and the child’s family:
Expected Outcome
The family will adhere to the medication regimen, as evidenced by normal thyroid hormone levels.
Expected Outcomes
The child will be euthyroid, as evidenced by normal results on thyroid function tests. The child will have normal bowel movements.
Expected Outcome
The child will be able to exercise at the same level as peers as basal metabolic rate returns to normal.
Expected Outcome
The child will sleep the appropriate amount of time for age as basal metabolic rate returns to normal.
Expected Outcome
The child will regain normal body temperature as basal metabolic rate returns to normal.
Interventions
The antithyroid drug PTU is usually given two or three times per day, whereas methimazole can be given once daily (LaFranchi, 2011a). A multiple-times-a-day medication regimen may be difficult for some children and parents to follow. Use of pill dispensers and a watch with an alarm to remind the child to take the medication at specific times enhances compliance. The endocrinologist should evaluate the child and monitor thyroid function every 2 to 4 months while the child is undergoing treatment. Normal values for thyroid function tests and alleviation of symptoms indicate appropriate responses to therapy.
Once the child is euthyroid and asymptomatic, the child should be evaluated once or twice a year. Medication dosages may be tapered after 2 to 3 years to evaluate for remission. Contact sports should be limited while the child is being treated to decrease the possibility of damage to the liver. Collaboration with the school nurse to facilitate medication administration is an important nursing function.
Evaluation
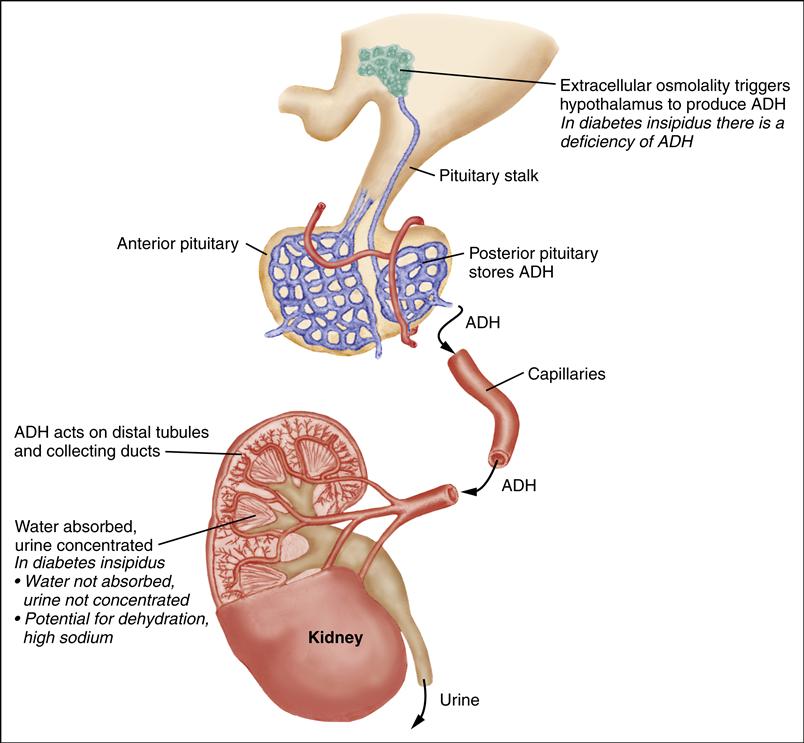
Diabetes Insipidus
Diabetes insipidus (DI) is an inability to concentrate urine. In central DI, there is a deficiency of vasopressin, also known as antidiuretic hormone (ADH). In nephrogenic DI, the kidneys are insensitive to vasopressin (Breault & Majzoub, 2011a).
Etiology
Both forms of DI can occur from inherited defects or acquired conditions (Breault & Majzoub, 2011a). Central DI frequently results from head trauma, tumors, or infection in the area of the hypothalamus. The most common type of tumor involving the hypothalamus that causes DI is craniopharyngioma. Cranial radiation for treatment of tumors may lead to ADH deficiency. Other causes include infections of the CNS such as meningitis or encephalitis, and congenital malformations such as septo-optic dysplasia or isolated pituitary malformation or ectopy. Several genetic mutations in the vasopressin gene causing DI have been identified (Cooperman, 2010). DI may also be idiopathic.
Incidence
DI is not common in the United States, occurring in 1 of every 25,000 individuals. Head trauma and cranial surgery account for the largest percentage of cases of DI. Thirty percent of cases are classified as idiopathic (Cooperman, 2010).
Manifestations
Increased urination (polyuria) and excessive thirst (polydipsia) are the classic manifestations of DI. Other signs and symptoms include nocturia and dehydration (Box 51-2).
Diagnostic Evaluation
Diagnostic criteria include polyuria with associated hypernatremia (greater than 150 mEq/L) and low urine specific gravity (less than 1.005) in the absence of hyperglycemia. Urine should be checked for glucose to rule out hyperglycemia as a cause of increased urine output.
A water deprivation test may also be necessary to confirm the diagnosis. In this 7- to 8-hour procedure, the child is deprived of all fluid intake. A normal response is decreased urine output with a high urine specific gravity and no change in serum sodium. In DI, when fluid is restricted, the child continues to have large amounts of dilute urine (low urine specific gravity). The serum sodium level also increases. To ensure the child’s safety, this test is done in a hospital setting with frequent monitoring of serum sodium, hematocrit, and osmolality. Urine osmolality and output are also measured. The child is weighed at the beginning, middle, and conclusion of the water deprivation test. Water deprivation should be stopped if the child loses 3% to 5% of baseline body weight, becomes dehydrated, or demonstrates a significant change in vital signs or neurologic status.
Therapeutic Management
Treatment for central DI involves maintaining fluid balance and administering synthetic vasopressin (1-deamino-8-D-arginine-vasopressin [DDAVP]). The dose of DDAVP ranges from 5 to 30 mcg/day (intranasal) or 2 to 4 mcg/day (intravenous [IV]/subcutaneous), divided into one or two doses per day. It is generally administered either intranasally, through a soft, flexible tube (rhinal tube) or metered spray, or by subcutaneous injection. The concentration of intranasal DDAVP is 100 mcg/mL; the concentration of subcutaneous DDAVP is 4 mcg/mL. The oral form of DDAVP is used primarily for nocturnal enuresis, although it has also been used in the treatment of DI. Oral doses range from 25 to 300 mcg every 8 to 12 hours (Cooperman, 2010).
Dosage is individualized on the basis of the child’s age, size, urine output, and urine specific gravity. The duration of action varies from 8 to 24 hours. Doses are timed so that before the next dose, the child is allowed to have mildly increased urination. This helps prevent overtreatment and water retention. Parents are often taught to measure urine specific gravity at home to monitor effectiveness of treatment.
 NURSING QUALITY ALERT
NURSING QUALITY ALERT
Diabetes Insipidus
The goals for the management of nephrogenic DI are to prevent severe dehydration and provide adequate calories for growth. With acquired disease, treatment is focused on eliminating the underlying cause; congenital nephrogenic DI is very difficult to treat (Breault & Majzoub, 2011a).
Nursing Considerations
Nursing care involves assessing the understanding of the child and parents about DI. The nurse educates the family about the basic pathophysiology of water metabolism and the cause of DI. This includes a description of signs and symptoms (increased thirst, polyuria, dehydration) and how they indicate the need for DDAVP, as well as signs and symptoms of excessive DDAVP administration (decreased urine output, headaches, water retention). The child must be closely monitored for signs