The Child with a Hematologic Alteration
Learning Objectives
After studying this chapter, you should be able to:
• Describe the anatomy and physiology of the hematopoietic system.
• Discuss the pediatric differences related to blood and blood formation.
• Discuss the role of the nurse in the prevention of iron deficiency anemia.
• Describe common factors in the care of a child with anemia.
• Discuss the pathophysiology and therapeutic management of common hematologic alterations.
• List possible nursing diagnoses for children with hematologic alterations.
• Describe possible nursing care for children with hematologic alterations.

http://evolve.elsevier.com/McKinney/mat-ch
CLINICAL REFERENCE
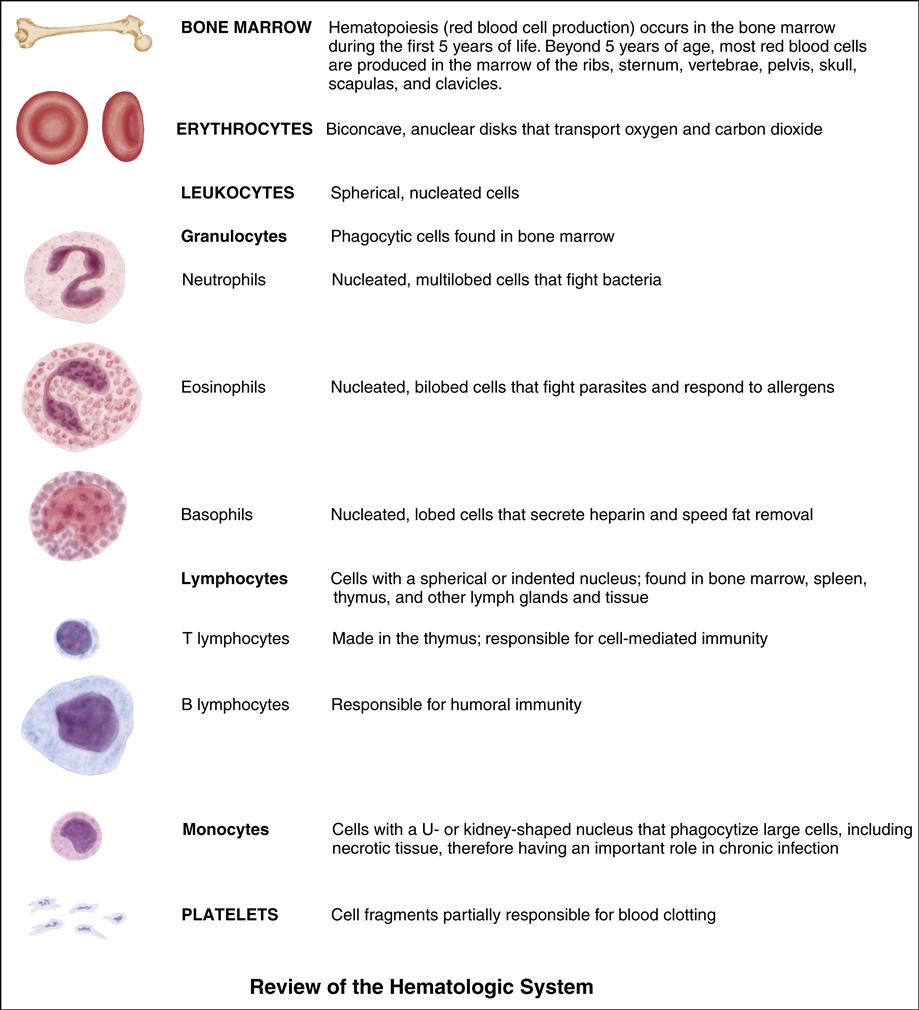
Review of the Hematologic System
Hematology is the study of the blood and blood-forming tissues. In fetal life, various tissues produce erythrocytes (red blood cells), but after birth, their production is controlled exclusively by the bone marrow, primarily in the long bones. With age, the more membranous bones of the vertebrae, sternum, and ribs assume red blood cell (RBC) production. Age, sex, and the altitude at which a person lives affect the number of RBCs.
Normally, RBCs are biconcave disks that are capable of changing shape as they flow through the microvasculature of the body. They have a fairly uniform size that is determined mainly by the amount of cellular content of substances, primarily hemoglobin. Their function is to transport oxygen to tissues. Essential to this ability to carry oxygen is an appropriate amount of hemoglobin, whose production depends on sufficient amounts of circulating iron. Iron is absorbed from dietary intake by the intestines and stored by the liver in both soluble and insoluble forms, to be used when necessary.
The stimulus for production of RBCs is a decrease in circulating oxygen, which in turn stimulates the kidneys to produce a hormone called erythropoietin. Erythropoietin stimulates the
production of RBC precursors and causes them to mature rapidly. Disorders of the kidney can affect the individual’s ability to produce this hormone and thus can affect RBC production by the bone marrow.
Anemia is a decrease in the number of RBCs, reduction in their hemoglobin content, or reduced volume of packed RBCs. Anemia results from one of two problems; either too rapid a loss of RBCs (by covert or overt bleeding or destruction) or too slow a production of RBCs. Anemias are categorized according to the size of the RBC (macrocytic, microcytic, normocytic) and the content of hemoglobin in the RBC (hypochromic, normochromic).
Polycythemia, which is an increase in the number of RBCs, is less frequently seen than anemia. Polycythemia can occur as a result of hypoxia, such as that experienced at high altitude or when oxygen is not sufficiently directed to the tissues, as in cyanotic heart disease.
White blood cells (WBCs), or leukocytes, are formed in the bone marrow and in lymphatic tissue. They assist in the body’s ability to distinguish “self” from “nonself.” WBCs destroy foreign cells through the processes of phagocytosis and antibody production. Both phagocytes and antibodies destroy foreign cells and tissues perceived by the body as nonself (including, e.g., bacteria, fungi, viruses, parasites, and transplanted tissue) (see Chapter 42). WBC disorders result from an altered rate of production of WBCs (lymphocytosis or lymphopenia) or an alteration in function of the cells.
Platelets are the cells that promote hemostasis—the prevention of blood loss. They are formed in the bone marrow from megakaryocytes. Megakaryocytes later fragment into smaller cells known as platelets, either in the bone marrow or shortly after release into the systemic circulation. Platelets can circulate in the blood for about 10 days before they die; however, disease, fever, and infection can shorten a platelet’s lifetime. Platelet disorders occur when the bone marrow cannot meet the production demands of the body.
When caring for infants and children with blood disorders, the nurse is challenged in the areas of preventive, acute, and chronic care. Depending on the disorder and the child’s condition, care may be provided in the home, an outpatient setting, or the hospital. It is not unusual for a child to be seen in an outpatient setting (clinic, school), referred to a hospital for diagnosis and stabilization, and returned to the home for maintenance. Genetic counseling may be indicated for children or families with certain types of blood disorders.
Many medications for hematologic disorders can be given at home, including deferoxamine mesylate (Desferal), intravenous immune globulin (IVIG), intravenous (IV) antibiotics, coagulation factor products, and, in some instances, even blood transfusions. Parents are learning to manage infusion therapy in regard to initiating, monitoring, and discontinuing infusions when appropriate, with guidance from the collaborative efforts of the multidisciplinary health care team.
Iron Deficiency Anemia
Iron deficiency is the most common cause of anemia during infancy, childhood, and adolescence. Iron deficiency anemia (IDA) may be characterized by mild or marked anemia.
Etiology and Incidence
Several factors can contribute to IDA, including decreased iron intake, increased iron or blood loss, and periods of increased growth rate.
IDA related to inadequate dietary iron intake is rare before age 4 to 6 months because of the presence of maternal iron stores; it occurs most often in children age 9 to 24 months as iron stores are depleted. Premature infants may develop iron deficiency early in life because they are born with insufficient maternal iron stores (Lerner & Sills, 2011). Decreased iron intake is often related to the intake of large amounts of cow’s milk instead of breast milk or fortified formula and iron-fortified foods (Lerner & Sills, 2011). The incidence has dropped in recent years because of increased education about and availability of iron-fortified formula and cereals. Early transition from breast milk or infant formula to cow’s milk can precipitate chronic diarrhea with occult intestinal bleeding in children younger than 2 years. This results from exposure to a protein found in cow’s milk (Lerner & Sills, 2011). The rapid growth of infants and children younger than 2 years, if combined with decreased iron intake, further contributes to the increased incidence in this age-group.
Adolescents are also at risk for IDA because they too are undergoing increased growth and often have poor dietary habits. The situation is further complicated by the blood loss during menstruation in young women.
Manifestations
The clinical manifestations of IDA vary with the degree of anemia but may include extreme pallor with porcelain-like skin, pale mucous membranes and conjunctiva, tachycardia, tachypnea, lethargy, fatigue, and irritability. Children with lead poisoning often have associated IDA.
Diagnostic Evaluation
Any child with anemia should first have a complete history taken, with particular emphasis on assessment of nutritional intake. The results of a complete blood count (CBC) in individuals with IDA will show low hemoglobin levels (6 to 11 g/dL) and microcytic, hypochromic RBCs, reflected in a decreased mean cell volume (MCV) and decreased mean cell hemoglobin (MCH). The reticulocyte count is usually normal or slightly elevated. With these findings, serum ferritin levels and serum iron or iron-binding capacity should be assessed. Total iron-binding capacity (TIBC) is usually increased as a result of decreased serum iron levels. Serum ferritin and serum iron are low. Hemoglobin electrophoresis may be done to rule out causes other than IDA.
Therapeutic Management
Therapy is directed toward increasing the dietary intake of iron and iron supplementation. The absorption of dietary iron-rich foods can be unreliable and will not rapidly provide the body with enough iron to correct the iron deficiency. Therefore, affected children are given a daily oral iron preparation (often three times per day) of one of the available ferrous salts (ferrous sulfate, ferrous gluconate, or ferrous fumarate) on the basis of the content of elemental iron (dose should be 3 to 6 mg/kg/day [2 to 4 mg/kg/day for premature infants] in three divided doses).
Follow-up monitoring includes a CBC and reticulocyte count. The reticulocyte count should increase within days of the initiation of iron therapy. An increased hemoglobin level can be expected in 4 to 30 days. The response to iron therapy can often
 NURSING QUALITY ALERT
NURSING QUALITY ALERT
Obtaining a Dietary Intake History
Parents may not readily or accurately respond to questions about their child’s dietary intake. Ask the parents to begin the dietary history at the time the child awoke yesterday, describing the child’s activities and exactly what the child ate. Correlating activities with diet may enable the nurse to obtain a better history and may alert the nurse to feeding patterns for which counseling may be indicated. Determine the number of bottles of milk or juice that the child is taking each day and the size of the bottles. Parents often underestimate the ounces of milk or juice that the child is taking.
Children with iron deficiency anemia occasionally develop pica. Pica is an appetite for nonnutritive substances such as paper, cardboard, ice, or sometimes dirt. Ask about the intake of nonfood items, because parents may not openly share this information.
 NURSING CARE PLAN
NURSING CARE PLAN
The Child with Iron Deficiency Anemia in the Community Setting
Focused Assessment
• Obtain history from parents addressing the following:
• Decrease in child’s activity level—more quiet than usual
• Infant given cow’s milk before age 12 months
• Increased milk consumption with exclusion of solid foods for infant and child
• Child’s heart “races” when at rest
• Conduct a physical examination and look for:
• Pallor
Nursing Diagnosis
Planning
Expected Outcome
The parents will have an understanding of the child’s nutritional needs, as evidenced by verbal description of the child’s dietary plan, including foods containing appropriate dietary iron.
Interventions and Rationales
1. Obtain the child’s past and current nutritional history. Instruct the caregiver to continue to give the infant iron-fortified formula or breastfeed and give supplementary iron-fortified foods until age 12 months. For a child older than 12 months, instruct parents to limit cow’s milk intake to a maximum of 24 oz/day and increase consumption of iron-rich foods.
Cow’s milk is poorly digested and is not rich in iron. The American Academy of Pediatrics (AAP) recommends continuing breast milk or iron-fortified formula until age 12 months. Decreasing milk intake will encourage the consumption of other iron-rich and iron-fortified foods (see Patient-Centered Teaching: Home Care of the Child with Iron Deficiency Anemia).
2. Advise the parent to keep a dietary diary.
Keeping a record of food and fluid intake assists with a dietary evaluation and identifies areas for additional teaching.
3. Explain to parents the need for iron in the manufacture of red blood cells (RBCs), how iron therapy affects laboratory test results, the potential outcome with no intervention, the lack of iron in cow’s milk, and iron’s effect on the body.
Explaining the rationale for therapy can often help improve adherence.
4. Instruct the parents to administer oral iron supplements as ordered by the physician. Allow parents to practice medication administration, and then evaluate their technique, providing additional instruction as indicated (see Patient-Centered Teaching: Home Care of the Child with Iron Deficiency Anemia).
Iron supplements immediately increase iron intake beyond that absorbed from formula or food. Practice and additional instruction will build the parents’ skills and confidence in giving their child medication.
5. Inform parents to give iron supplements with fruit juice and NOT with milk, formula, or cereal; when the child’s stomach is empty.
An acid stomach environment facilitates absorption. Calcium in milk products binds with iron to decrease iron absorption.
6. Instruct parents to administer vitamin C as ordered and encourage intake of foods rich in vitamin C.
Vitamin C increases the absorption of iron by the body.
7. Instruct the parents to keep iron supplements (and all medications) out of reach of children and in a locked cabinet.
Iron poisoning is possible with overdose. This can be serious and possibly fatal.
8. Instruct the parents to obtain follow-up laboratory tests when ordered, including reticulocyte count and hemoglobin and hematocrit levels.
The reticulocyte count should peak in 5 to 7 days. It is an objective test that can determine the degree of adherence to iron therapy. The hemoglobin level should increase in 4 to 30 days.
9. Instruct the parents to monitor for and expect to see black, tarry stools. Verify that parents have observed this.
Iron therapy causes black, tarry stools. If absent, it may indicate lack of adherence to therapy.
10. Refer to social services for enrollment in federal or state programs, if warranted.
Poor nutritional practices may be attributable to a lack of resources to obtain appropriate foods.
Evaluation
be positively predicted, so blood transfusions are rarely indicated to correct IDA. RBC transfusions are reserved for severe anemia and cardiovascular compromise. Oral iron therapy will be continued for 3 to 4 months after hemoglobin and hematocrit levels return to normal, in order to replenish the child’s iron stores. Then administration of a daily multivitamin with iron can be recommended.
Sickle Cell Disease
Sickle cell disease (SCD) is the generic term that refers to a group of genetic disorders characterized by the production of sickle
 CRITICAL THINKING EXERCISE 47-1
CRITICAL THINKING EXERCISE 47-1
Mrs. Anders has brought 18-month-old Jacob to the clinic because he is irritable, is running a low-grade fever, and has a cough. In the process of assessing Jacob, you note that he seems lethargic and his skin is very pale. You obtain a 24-hour diet history and suspect the child does not have adequate iron intake. A complete blood count (CBC) confirms a diagnosis of iron deficiency anemia (IDA).
hemoglobin (HbS), chronic hemolytic anemia, and ischemic tissue injury. The more common forms of SCD include homozygous sickle cell disease (HbSS) or sickle cell anemia, sickle hemoglobin C disease (HbSC), and the sickle beta-zero thalassemia syndromes. SCD is an inherited, lifelong disease that affects primarily individuals of African, Mediterranean, Indian, and Middle Eastern descent. The morbidity and mortality rates from the severe forms of the disease have decreased as a result of newborn screening for the disease, routine prophylactic penicillin administration, and pneumococcal and Haemophilus influenzae vaccines.
PATHOPHYSIOLOGY
Sickle Cell Disease
The normal red blood cell (RBC) is a smooth, biconcave disk that is capable of changing shape to enable it to flow easily through the microvasculature of the circulation. Under conditions of low oxygen concentration, acidosis, and dehydration, the RBCs in a child with sickle cell disease (SCD) assume a sickle shape similar to a half moon, which prevents them from flowing easily through the smallest blood vessels. Sickled RBCs are stiff and nonpliable. These sickled cells clump together, causing occlusions in the small vessels. With reoxygenation, most of the sickled RBCs resume their normal shape. After repeated sickling (deoxygenation) and unsickling (reoxygenation), the cells become irreversibly sickled, and their life span is reduced from 120 days to 12 days.
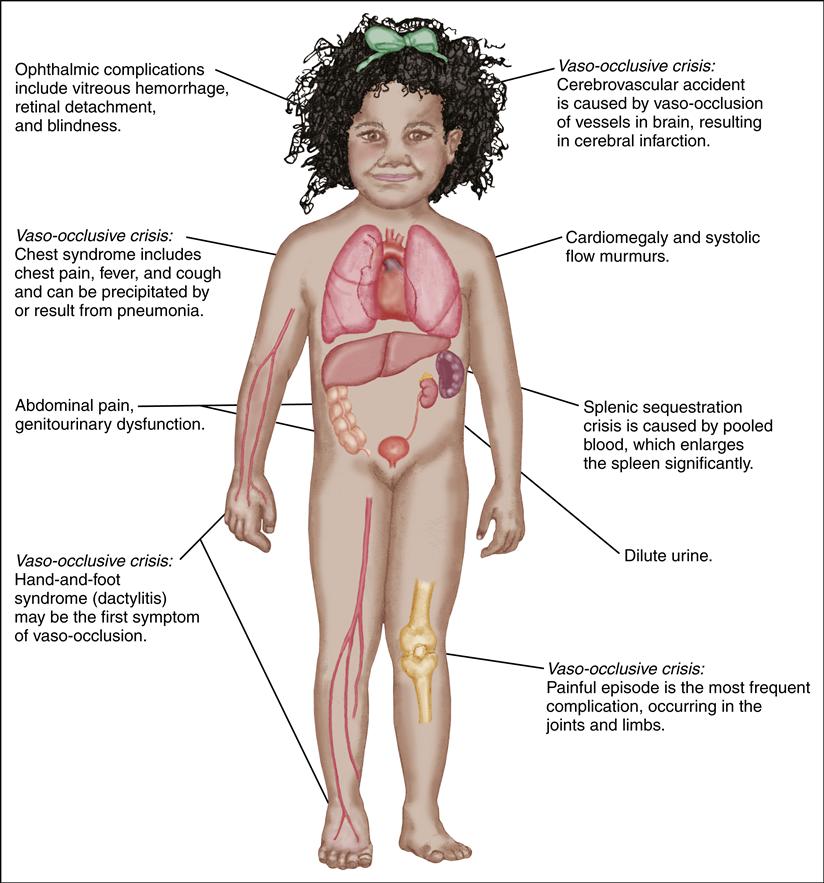
Sickled cells cause microvascular occlusion, leading to tissue ischemia, infarcts, and organ damage. The lungs, spleen, and brain are the organs most seriously affected by the complications of SCD. The normal spleen functions to filter bacteria in the blood. However, the spleen of a child with SCD does not function properly much beyond age 5 years because of this repeated microvascular occlusion and damage. The large vessels are also affected, leading to strokes and other vaso-occlusive events.
Etiology
SCD is a group of hemoglobinopathies in which normal hemoglobin A, (HbA), is partially or totally replaced by abnormal hemoglobin, HbS. SCD is an inherited, autosomal recessive condition. Each affected individual carries two copies of the gene that directs the production of hemoglobin. The normal hemoglobin gene is for HbA. If one parent has the HbS trait (one copy of the HbA and one copy of HbS gene—HbAS) and the other parent does not (both copies HbA—HbAA), each pregnancy has a 50% risk of having the child inherit the trait. If each parent carries the trait (both parents with HbAS), there is a 25% chance that the child will be unaffected (HbAA), a 50% chance that the child will carry the trait (HbAS), and a 25% chance that the child will have the disease (HbSS). Although not a disease in and of itself, the carrier state of SCD—sickle cell trait (HbAS)—may produce clinical symptoms (e.g., hematuria, bacteriuria) in times of extreme stress, during extremely vigorous exercise, and at high altitudes.
Incidence
SCD affects approximately 1 in 500 African-Americans. Sickle cell trait is found in 8% of persons of African descent and is also prevalent in persons of Mediterranean, Middle Eastern, Indian, Caribbean, and Central and South American descent (Heeney & Dover, 2009).
Manifestations
All the clinical manifestations of SCD are a result of the obstructions caused by the sickled RBCs and the increased destruction of RBCs because of their shortened life span in SCD. Large amounts of fetal hemoglobin (HbF) present in the first few months of life obscure the presence of HbS, so symptoms of the disease usually do not appear until age 4 to 6 months, when the infant begins to manufacture hemoglobin S.
The disease affects most organ systems. Delayed growth and puberty are common. The child usually has small stature throughout adolescence but may attain normal growth in the early 20s.
The general manifestations of SCD are chronic hemolytic anemia, pallor, jaundice, fatigue, cholelithiasis, delayed growth and puberty, avascular necrosis of the hips and shoulders, renal dysfunction, and retinopathy. Sickling events may also progress to acute episodic exacerbations known as sickle cell crisis. Infection, dehydration, hypoxia, trauma, or general stress may precipitate a crisis episode. The crisis may take one of three forms: vaso-occlusive, acute sequestration, or aplastic.
A vaso-occlusive crisis occurs when blood flow to tissues is obstructed by sickled RBCs, leading to hypoxemia and ischemia. This ischemic event causes the child to experience pain in the affected body part.
An acute sequestration event occurs when blood flow from an organ, such as the liver, lungs, or spleen, is obstructed by sickled RBCs. These organs then become engorged with blood, leading to acute anemia and, when in the lungs, acute chest syndrome with potential respiratory failure.
An aplastic event occurs when there is either an increased destruction or decreased production of RBCs. Increased destruction can be related to fever or infection. Decreased production is frequently associated with a viral infection such as parvovirus B19 (also known as Fifth disease).
Repeated vaso-occlusive crises and a virtually continual state of anemia produce long-term problems later in the life of the individual with SCD. Table 47-1 presents the clinical manifestations of SCD.
TABLE 47-1
CLINICAL MANIFESTATIONS AND THERAPEUTIC MANAGEMENT OF SICKLE CELL DISEASE COMPLICATIONS
| COMPLICATION | CHARACTERISTICS | MANIFESTATIONS | TREATMENT |
| Vaso-Occlusive Crisis | |||
| Painful episode | Most common type of crisis and reason for hospitalization Typically produces bone or joint pain, but pain can occur anywhere Pain may come and go Frequency of pain is individualized Pain is precipitated by infection, cold, stress, acidosis, local or generalized hypoxia | Mild: Joint or bone pain lasting a few hours Severe: Joint or bone pain lasting days | Oral analgesics initially and, if ineffective, IV opioids (usually morphine), which may be given by either intermittent or continuous infusion Oral or IV NSAIDs Oral and IV hydration Aggressive incentive spirometry use (10 breaths every 2 hr when awake) Consistent manner to assess subjective experience of pain is essential Use of nonpharmacologic pain management strategies in addition to medications |
| Acute chest syndrome | Common cause of hospitalization Sometimes confused with pneumonia Can recur | Chest pain, fever, cough, abdominal pain | IV hydration (1-1½ times maintenance), antibiotics, oxygen, RBC transfusion, analgesics |
| Dactylitis (hand-and-foot syndrome) | Occurs in children ages 6 mo to 4 yr Self-limiting complication | Swelling of hands or feet, pain, warmth in affected area | Oral analgesics, hydration (oral or IV), rest |
| Priapism (persistent erection of the penis) | Occurs if penile blood flow becomes obstructed | Persistent, painful erection | Analgesics; hydration Avoid hot and cold packs If prolonged, transfusion therapy |
| Cerebrovascular accident | Without treatment, mortality rate of 20%; 70% of patients have a recurrence | Hemiparesis or monoparesis, aphasia/dysphasia, seizures, alteration in level of consciousness, vomiting, vision changes, ataxia, headache | Long-term RBC transfusion therapy or erythrocytapheresis and possibly chelation therapy May require extensive rehabilitation |
| Acute Sequestration Crisis | |||
| Blood volume pooling in the spleen, causing splenic enlargement Life-threatening condition of hypovolemic shock Usually occurs in children ages 6 mo to 4 yr One episode increases the risk of future occurrences | Decreased hemoglobin level, acutely ill-looking child, pallor, irritability, tachycardia, impressively enlarged spleen, hypovolemic shock | Emergency treatment to restore circulating blood volume with crystalloid and colloid (blood) infusion Long-term transfusion therapy if recurrent Eventual splenectomy in cases of persistent recurrence | |
| Aplastic Crisis | |||
| Profound anemia caused by diminished erythropoiesis Has been observed after parvovirus-like agent exposure | Pallor, lethargy, headache, fainting | RBC transfusions and treatment of symptoms | |
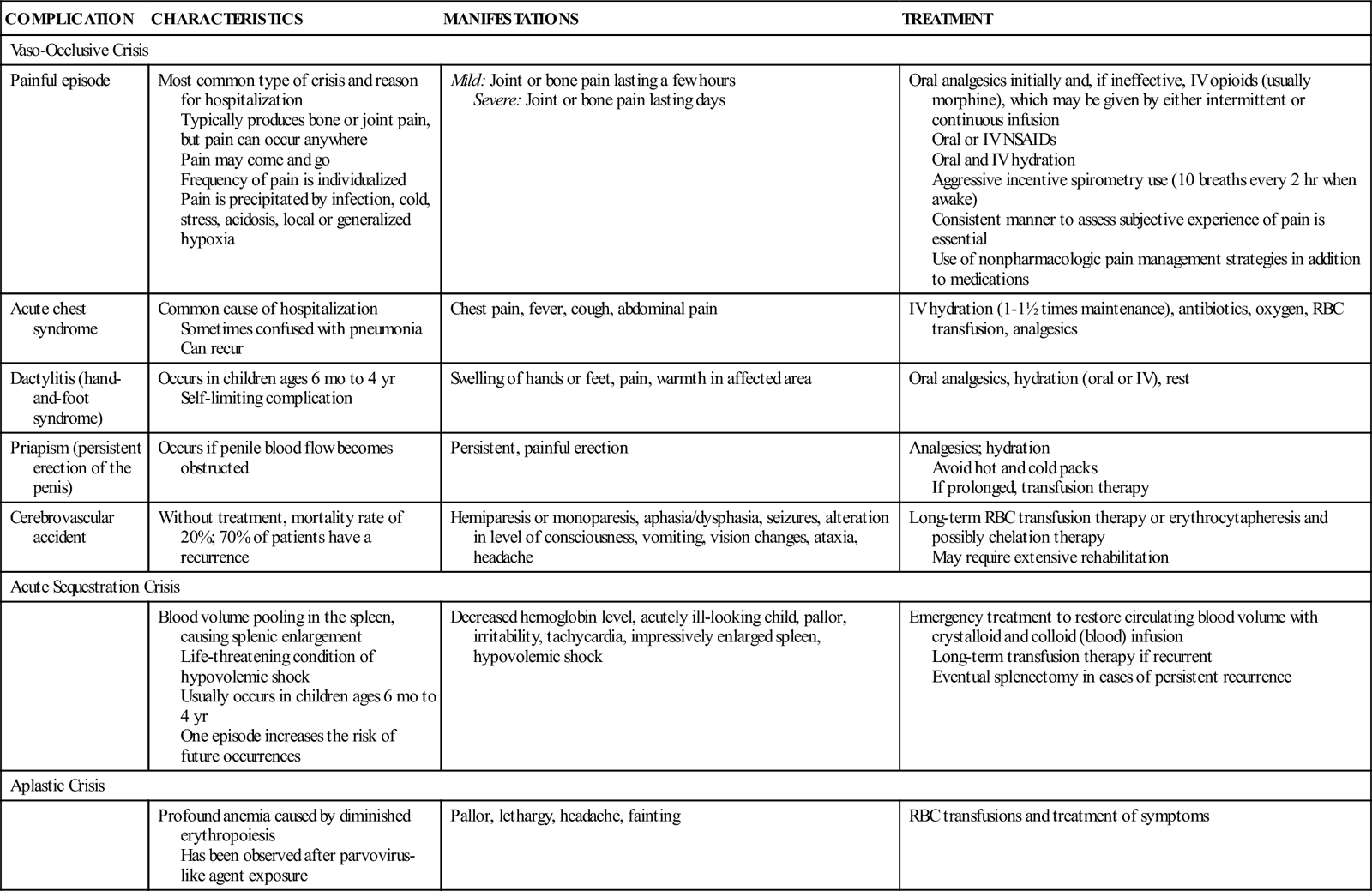
IV, Intravenous; NSAIDs, nonsteroidal antiinflammatory drugs; RBC, red blood cell.
Diagnostic Evaluation
In the past, many infants died from complications of SCD before being diagnosed with the disorder. However, newborn screening for SCD has significantly decreased the mortality rate. A laboratory diagnosis of SCD is established on the basis of a CBC, isoelectric focusing, hemoglobin electrophoresis, and high-performance liquid chromatography. Children with SCD have elevated reticulocyte counts because of the shortened life span and destruction of sickled RBCs. Prenatal diagnosis is an option and is made by chorionic villus sampling at 8 to 10 weeks of gestation or amniocentesis at 15 weeks of gestation.
Therapeutic Management
In SCD, the spleen often does not function properly or has been surgically removed because of complications. Functional or actual asplenia places children and adults with SCD in an immunocompromised state at high risk for infection. Splenic dysfunction can begin at 6 months of age, and those with HbSS may have total dysfunction by age 5 years. Bacterial septicemia poses a severe health risk to children with SCD (DeBaun, Frei-Jones, & Vichinsky, 2011a). The bacteria Streptococcus pneumoniae and H. influenzae are normally destroyed by the reticuloendothelial system of the spleen, but because the child with SCD does not have a properly functioning spleen, the child is considered to be more susceptible to infection with these bacteria.
The natural history of splenic dysfunction in children with HbSS places them at higher risk for fulminant septicemia and death during the first 3 years of life than children with HbSC. Prophylactic daily penicillin V therapy (given twice a day) is recommended for all children with suspected or actual diagnosis by age 2 months and is continued until at least age 5 years (American Academy of Pediatrics [AAP] Committee on Infectious Diseases, 2009). Although views regarding the use of penicillin differ, some experts will continue penicillin prophylaxis throughout childhood in high-risk patients with asplenia.
The possibility exists that pneumococcal infections will not be completely prevented by penicillin chemoprophylaxis because of the development of penicillin-resistant pneumococcal strains (AAP Committee on Infectious Diseases, 2009). The pneumococcal polyvalent vaccine is routinely recommended for children at ages 2, 4, 6, and 12 to 15 months of age (AAP Committee on Infectious Diseases, 2009). Routine vaccination against H. influenzae infections and routine immunization against hepatitis B are recommended as well. Moreover, children with SCD should receive the influenza vaccine annually and immunization against meningococcal disease after age 2 years (AAP Committee on Infectious Diseases, 2009) because of their increased risk for related complications.
Stroke occurs in 7% to 13% of children with the HbSS form of SCD. Stroke, caused by vaso-occlusion of blood vessels in the brain with sickled RBCs leading to ischemia, can result in significant motor and neuropsychological deficits. The goal of treatment is early identification of those at increased risk and intervention to prevent the occurrence of a stroke. The risk of stroke can be predicted by the performance of a transcranial Doppler ultrasound. Those with abnormal results are often managed to reduce their risk of stroke by either monthly transfusions or erythrocytapheresis. By maintaining the child’s hemoglobin S at less than 30% with the transfusion of hemoglobin A−containing RBCs, the marrow’s production of additional hemoglobin S can be suppressed, therefore reducing the risk of stroke (Mazumdar, Heeney, Cox, et al., 2007).
A child with SCD and a temperature of 101.3° F (38.5° C) or higher should receive prompt medical evaluation and treatment because of the overwhelming risk for infectious complications. Fever can be the first sign of bacteremia. Parenteral antibiotics, blood cultures, IV hydration, and general monitoring are the standard of care for children with SCD who have fever. Outpatient therapy with long-acting parenteral antibiotics may be provided in combination with rigorous evaluation and follow-up in those centers with the capabilities to do so. Children who are not eligible for outpatient therapy are those with the following signs and symptoms (and thus they are considered at high risk for sequelae):
• Prior splenectomy or history of pneumococcal sepsis
• Family’s or child’s lack of ability to adhere to outpatient therapy
• WBC count less than 500/mm3 or more than 30,000/mm3
Opioids and nonsteroidal antiinflammatory drugs (NSAIDs) (i.e., ibuprofen, ketorolac) are the mainstays of analgesic treatment, particularly in combination for painful crises. Morphine is the current opioid of choice. Meperidine (Demerol) is not recommended for long-term pain management because of its side-effect profile. Opioids provide systemic relief, and the NSAIDs act locally to decrease inflammation at the site of vaso-occlusion and provide analgesia without the potential side effect of respiratory depression. Morphine has been very effective when administered intravenously, particularly when given using the patient-controlled analgesia method (see Chapter 39).
 NURSING CARE PLAN
NURSING CARE PLAN
The Child with Sickle Cell Disease
Focused Assessment
• On initial diagnosis, obtain history related to parents’ concern that their child is in pain. Ask if the child:
• Refuses to move an extremity
• Cries out when a joint is moved or touched
• Assess for possible causes of pain other than SCD.
• Assess the severity of the child’s condition.
• When assessing pain in adolescents with SCD, consider the following:
• They want to fit in with their peer group, but they are different because of SCD.
• Obtain objective data specific to the type of painful episode.
Nursing Diagnosis
Planning
Expected Outcomes
Interventions and Rationales
1. Monitor the child’s vital signs and respiratory status every 4 hours and as needed.
Vital signs and respiratory status are assessed frequently to detect changes in tissue perfusion and respiratory status. Signs of altered perfusion include increased respiratory rate, increased work of breathing, decreased oxygen saturation, poor color, mottled appearance, prolonged capillary refill time, decreased peripheral pulses, and altered level of consciousness. A change in the level of consciousness often indicates poor perfusion or oxygenation of the brain.
2. Monitor oxygen saturation using pulse oximetry and administer oxygen to keep saturation greater than 95%.
Ensuring adequate oxygen can ease the child’s work of breathing and facilitate tissue oxygenation. Oxygen does not reverse the sickling process but may prevent more sickling.
3. Ensure adequate hydration by measuring intake and output and administering crystalloids and colloids (red blood cells [RBCs]) as ordered.
Measuring intake and output monitors renal function and level of hydration. Transfusions of crystalloids and (nonsickled) RBCs will increase the oxygen-carrying capacity of the blood and decrease the relative number of sickled cells.
4. Determine the child’s and family’s understanding of SCD and the treatment regimen and provide information or clarification as necessary.
Adequate understanding of SCD can increase adherence to preventive measures and prompt interventions during exacerbations and hospitalizations.
Evaluation
Nursing Diagnosis
Planning
Expected Outcome
The child will have decreased pain, as evidenced by a lowered score on the selected pain assessment tool.
Interventions and Rationales
1. Monitor and record pain level every 1 to 2 hours and more frequently if needed, using a pain assessment tool appropriate for the child’s age.
Pain can be severe in vaso-occlusive crisis and is relieved for only short periods. An assessment tool that measures the subjective experience of pain is helpful in determining the child’s level of discomfort (see Chapter 39).
2. Administer analgesics as ordered.
Analgesics may be administered intermittently, by a patient-controlled analgesic (PCA) pump (or PCA by proxy), and by continuous infusion through an intravenous (IV) line.
3. Increase oral fluids, if able to tolerate, or administer fluids IV at a rate that is 1 to 1½ times the maintenance rate.
Increased fluid volume reduces the viscosity of the blood, thus alleviating sites of vascular occlusion and preventing further sickling caused by dehydration.
4. Administer RBCs as ordered.
Maintaining an adequate hemoglobin level increases oxygen-carrying capacity to aid in further preventing sickling and microvascular ischemia.
5. Incorporate the use of age-appropriate, non-pharmacologic pain relief measures.
Comfort measures often help distract the child from discomfort (see Chapter 39).
6. Perform passive range-of-motion exercises and avoid exertion.
Passive range-of-motion exercises promote circulation without exacerbating fatigue.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


