The Child with a Gastrointestinal Alteration
Learning Objectives
After studying this chapter, you should be able to:
• Describe the anatomy and physiology of the gastrointestinal system in the infant and child.
• State expected nursing diagnoses for gastrointestinal alterations.
• Develop home care guidelines for the child with gastrointestinal alterations.
• Implement child and family teaching.
• Demonstrate critical thinking skills to manage a given patient care situation.

http://evolve.elsevier.com/McKinney/mat-ch
CLINICAL REFERENCE
Review of the Gastrointestinal System
Upper Gastrointestinal System
The upper gastrointestinal (GI) system includes the mouth, esophagus, and stomach. Its primary functions are to take in food and fluids, begin the digestive process, and propel food into the intestines, where nutrients are absorbed. The mouth, or buccal cavity, is the entrance to the GI tract. Here food is broken up and mixed with saliva. This process starts the digestion of carbohydrates. The submandibular, parotid, and sublingual glands secrete saliva in response to the smell, taste, or thought of food. The tongue contains taste buds that distinguish salt, sweet, sour, and bitter sensations. The tongue is essential for swallowing.
At birth, the esophagus measures approximately 10 cm in length; it lengthens to 18 to 25 cm by adulthood. The upper third of the esophagus consists of striated voluntary muscle; the lower two thirds consist of smooth muscle. The upper esophageal sphincter (UES) prevents the reflux of esophageal contents into the pharynx and lungs and prevents esophageal distention during respiration; the lower esophageal sphincter (LES, or cardiac sphincter) prevents the reflux of gastric contents into the lower esophagus.
Swallowing is under both voluntary and involuntary control. As food is chewed, it forms a small bolus, or mass; the tongue propels the bolus toward the oropharynx. The presence of this mass in the oropharynx stimulates the medulla, causing the soft palate to rise. The nasal passages close, the pharyngeal muscles contract, the larynx closes, and respiration is inhibited. As a
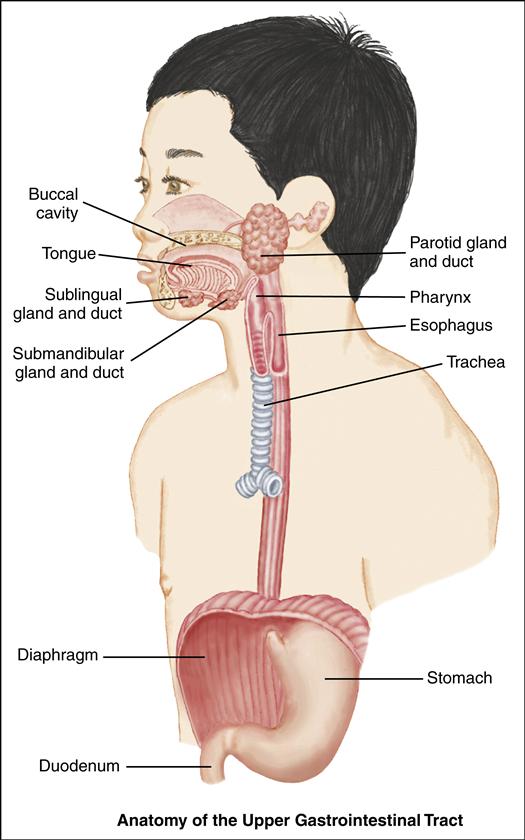
result of these processes, food is propelled to the esophagus. Through peristalsis, the bolus moves on to the LES, the muscle relaxes, and the bolus enters the stomach.
The stomach lies in the epigastric, umbilical and left hypochondrial regions of the abdomen. It is a muscular pouch, shaped somewhat like a gourd, where the bolus is received. As the LES and the pylorus contract, the stomach muscles churn the contents. The contents mix with the digestive juices to form chyme. The chyme moves on to the pylorus and into the duodenum.
A mucous-bicarbonate barrier in the stomach provides a thick layer of mucus and a buffer zone to neutralize acid. Stomach acids diffuse slowly through this layer toward the gastric wall. They are neutralized by bicarbonate ions from the surface epithelial cells. Thus a neutral pH is maintained at the gastric epithelial surface.
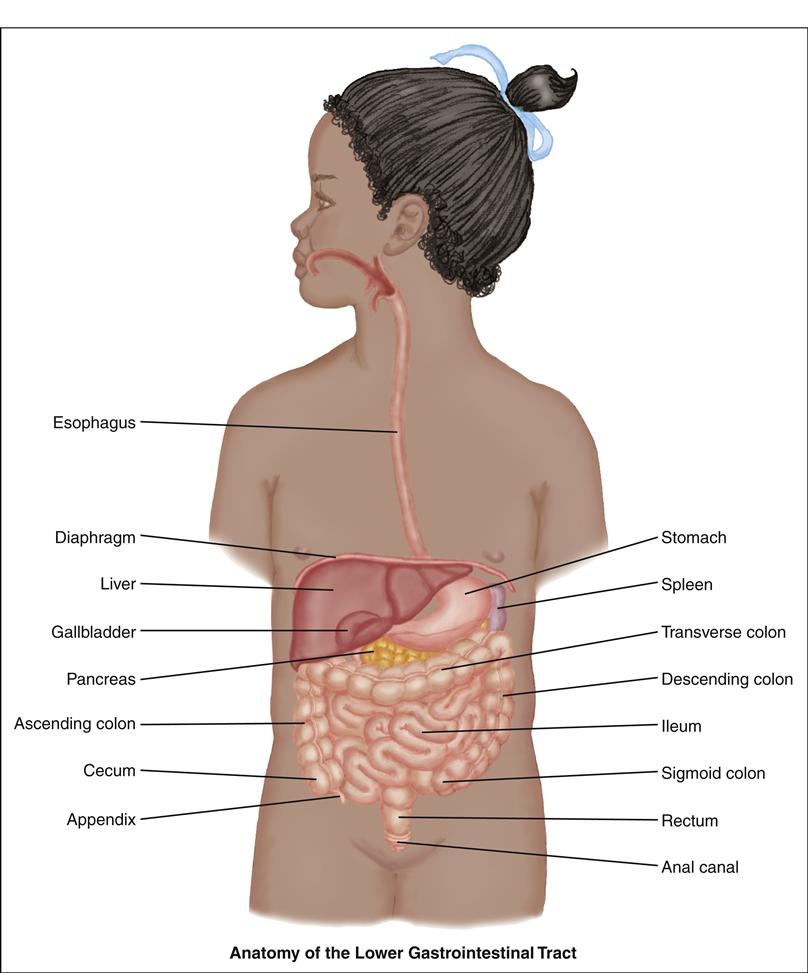
Lower Gastrointestinal System
The lower GI system includes the duodenum, liver, gallbladder, pancreas, jejunum, ileum, cecum, appendix, ascending colon, transverse colon, descending colon, sigmoid colon, rectum, and anus. The primary functions of the lower GI tract are to digest and absorb nutrients, detoxify and excrete unwanted waste, and aid in fluid and electrolyte balance.
The duodenum, the first part of the small intestine, extends from the pylorus to the jejunum. Partially digested chyme from the stomach enters the duodenum, where pancreatic enzymes and bile are excreted to further break down fats, carbohydrates, and proteins. The pancreas is an oblong gland lying behind the stomach that secretes enzymes to digest food and secretes glucagon and insulin to control motility and absorption.
The liver, the largest organ in the body, is located under the right diaphragm. The liver lies predominantly in the right upper quadrant, with the left lobe extending into the left upper quadrant. It is divided into two lobes separated by the falciform ligament. Within each lobe are numerous lobules, which form the functional units of the liver.
The liver is unique in that it is supplied with blood from two sources: (1) the hepatic artery, which supplies oxygenated blood; and (2) the hepatic portal vein, which supplies deoxygenated blood with absorbed nutrients from the GI tract. The liver has numerous functions, including phagocytosis, bile production, detoxification, glycogen storage and breakdown, and vitamin storage. The production of bile is essential for the absorption of fat and the excretion of the end products of blood cell breakdown. The primary function of the gallbladder, a saclike structure attached to the underside of the right lobe of the liver, is to store bile for secretion into the duodenum when stimulated by the presence of fat in its lumen.
The jejunum and ileum form the remainder of the small intestine. Absorption of all nutrients and vitamins occurs here through the villi and microvilli by the processes of diffusion and active transport. Absorption of vitamin B12 occurs only in the terminal ileum.
The large intestine starts with the cecum. This blind pouch, 2 to 3 inches long, begins at the ileocecal valve, which prevents reverse peristalsis into the small intestine. Attached to it is the appendix, a wormlike tube about 3 inches long. The open end of the cecum attaches to the remainder of the colon, which is divided into four sections: the ascending, transverse, descending, and sigmoid colon. One major function of the large intestine is water reabsorption, which occurs mostly in the cecum and ascending colon. Intestinal bacteria ferment the remaining carbohydrates and aid in the synthesis of vitamins B and K. Final breakdown of bile occurs here. Mucus secretion and peristalsis of wastes are also important functions.
The rectum is the last 7 to 8 inches of the intestine, and the anal canal refers to the last 1 to 2 inches. Stool is stored in the rectum until distention of the rectal walls initiates the defecation reflex—the final stage of the GI processes.
Prenatal Development
The primitive gut is formed from the endoderm in the first 4 weeks of embryonic development. The primitive gut then gives
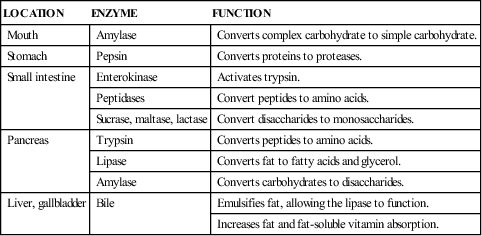
COMMON LABORATORY AND DIAGNOSTIC TESTS FOR GI DISORDERS
| TEST | DESCRIPTION | NORMAL FINDINGS | INDICATIONS | PREPARATION AND NURSING CONSIDERATIONS |
| Stool | ||||
| Culture and sensitivity | Organisms from a small sample of stool are grown in culture media. | Normal GI flora | To identify infectious organisms and determine their antibiotic sensitivity. | No patient preparation is necessary. The sample is delivered to the laboratory immediately; it must be kept free from contamination. |
| Reducing substances (Clinitest) | Stool is diluted with water and then tested for undigested carbohydrates with Clinitest tablets. | Negative | Used to diagnose malabsorption syndromes. | No preparation is necessary. The test is done by the nurse, who checks for a color change in the solution. |
| Occult blood (guaiac, Hematest) | Stool is smeared on filter paper and prepared with solution. | Negative | Used in inflammatory conditions and bowel necrosis. | No preparation is necessary. The test is done by the nurse; a blue color is positive. |
| Ova and parasites (O&P) | Stool is examined microscopically for presence of parasites or their eggs. | Negative | To identify enteric parasites in child with diarrhea or abdominal pain. | No patient preparation is necessary. Sample must be free from water or urine contamination. The sample is delivered to the laboratory either fresh or in preservatives. Barium, antacids, mineral oil, and antibiotics may interfere with results. 1-3 samples are collected. |
| Urine | ||||
| Urobilinogen | Dipstick or laboratory analysis is performed to determine bile byproducts in urine. | Negative | Levels determined in hepatic dysfunction and obstruction. | No preparation is necessary. The test is done by the nurse. |
| Blood | ||||
| Liver function tests | Serum levels are measured to give an indication of liver function. | AST: child < 9 yr, 15-55 U/L; child > 9 yr, 5-45 U/L ALT: 5-45 U/L Total bilirubin: 0.2-1.0 mg/dL Ammonia: 29-70 mcg/dL in children; 90-150 mcg/dL in newborns | Studies are performed when liver problems are suspected. | No preparation is necessary. Venipuncture is performed. |
| Endoscopy | ||||
| Fiberoptic upper GI endoscopy | Study allows direct viewing of the lining of the esophagus, stomach, and proximal duodenum. It also provides a means to obtain material for biopsies and cultures. | Normal mucosa | Used to rule out various upper GI tract disorders. | Preparation includes teaching, keeping the child on NPO status for at least 6 hr before the examination, providing conscious sedation, and monitoring the child’s respiratory function during sedation. |
| Colonoscopy | The colon is viewed directly by a fiberoptic scope and camera inserted rectally. | Normal mucosa and patent bowel | Performed to detect mucosal changes and abnormalities in the lumen of the colon. | Preparation includes teaching, keeping the child on NPO status, bowel cleansing, and providing conscious sedation. |
| Biopsy (gastric, jejunal, rectal, liver) | A small piece of tissue is removed for analysis. | No abnormal tissue | Study determines the amount of mucosal inflammation and the absence of ganglion cells. | Preparation includes teaching, bowel cleansing, and providing sedation or anesthesia if the procedure is done percutaneously (liver). |
| Radiologic Examinations | ||||
| Abdominal flat plate | Anterior and posterior radiographs are obtained. | Radiographs demonstrate stool and gas patterns, inflammation, and patency of the GI tract. It is commonly performed in cases of abdominal pain, imperforate anus, intussusception, and appendicitis. | Usually no preparation is necessary other than teaching. | |
| Barium swallow examination | Radiopaque contrast medium or air (or both) is swallowed. | Normal swallowing, no anatomic defects | Study identifies esophageal abnormalities, swallowing difficulties, and sphincter function. | Preparation includes teaching and keeping the child on NPO status for 2-4 hr before the examination. Adequate fluids are essential after the examination to prevent barium impaction. |
| Upper GI examination | Radiopaque contrast material is swallowed or inserted by NG tube. | Normal gastric emptying and no other abnormalities | Study outlines the stomach and pyloric canal and can be used to determine gastric emptying time. | Preparation includes teaching and keeping the child on NPO status for 4 hr before the examination. Adequate fluids are essential after the examination to prevent barium impaction. |
| Barium enema, air-contrast barium enema examination | Radiopaque contrast material or air (or both) is placed in the large intestine via the rectum. | Patent bowel and no abnormalities | Study used to identify abnormalities on the surface of the bowel lumen and to determine bowel patency. It also provides hydrostatic reduction of intussusception. | Preparation includes teaching, keeping the child on NPO status, and bowel cleansing. Adequate fluids are essential after the examination to prevent barium impaction. |
| CT scan | Oral radiopaque contrast material often used. May also use IV or rectal contrast. | Normal anatomy without evidence of inflammation | Used to identify inflammatory conditions and appendicitis. | No patient preparation other than teaching. Sedation may be used if child unable to remain still. Can be completed in less than 15 min. |
| Other | ||||
| Ultrasound | Study uses sound waves noninvasively to image anatomy and inflammation. | No abnormalities | Performed to identify anatomic abnormalities and inflammatory conditions. | Preparation includes teaching. For pelvic ultrasound, a full bladder is needed to improve imaging of pelvic organs. |
| Breath hydrogen test | Carbohydrate solution is given by mouth and exhaled. Breath samples are collected over 3 hr. | Less than 20 ppm above baseline | Used to diagnose maldigestion or malabsorption syndromes. Inadequately digested carbohydrate produces hydrogen when acted on by GI flora. | Nursing preparation entails teaching about the procedure. The child prepares by fasting for 4½ hr. The study is noninvasive. A facemask may be worn to collect expired air. |
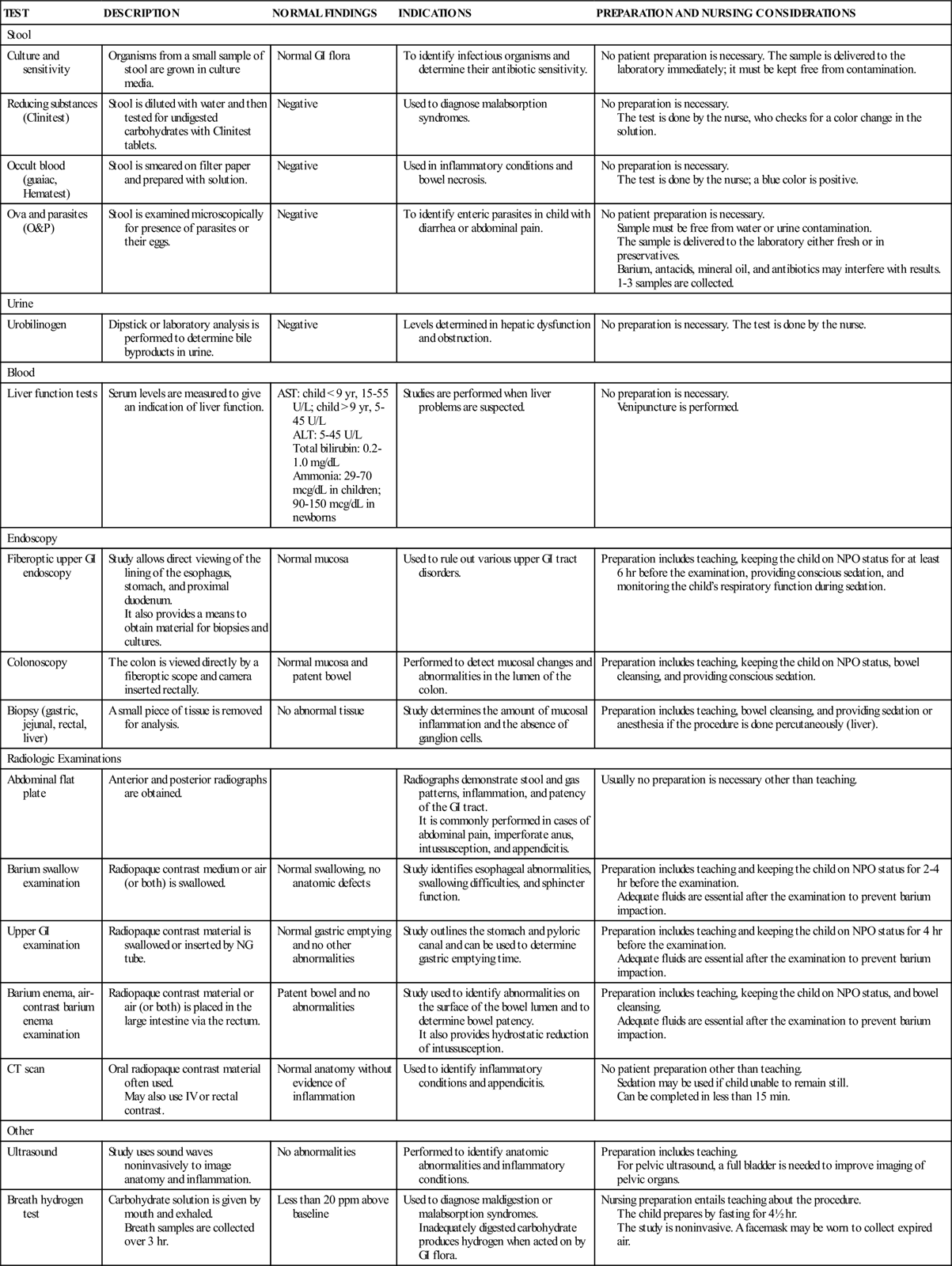
| LOCATION | ENZYME | FUNCTION |
| Mouth | Amylase | Converts complex carbohydrate to simple carbohydrate. |
| Stomach | Pepsin | Converts proteins to proteases. |
| Small intestine | Enterokinase | Activates trypsin. |
| Peptidases | Convert peptides to amino acids. | |
| Sucrase, maltase, lactase | Convert disaccharides to monosaccharides. | |
| Pancreas | Trypsin | Converts peptides to amino acids. |
| Lipase | Converts fat to fatty acids and glycerol. | |
| Amylase | Converts carbohydrates to disaccharides. | |
| Liver, gallbladder | Bile | Emulsifies fat, allowing the lipase to function. |
| Increases fat and fat-soluble vitamin absorption. |
rise to the following three sections of the GI tract, each having an individual blood supply and rate of development:
• Foregut: From the pharynx to the duodenum, including the liver, pancreas, and biliary tract
• Midgut: From the duodenum to the transverse colon
Problems in the development of each of these three sections give rise to specific malformations and disease states. Anatomically, development is complete at birth, but physiologically, the neonate’s GI tract is immature.
Fetal swallowing, intestinal motility, and defecation are detectable in the second trimester of gestation, but the most rapid and extensive development of the GI system occurs in the third trimester. The newborn must be able to adapt from total parenteral nutrition to total enteral nutrition because the placenta no longer performs nutrient exchange and waste removal.
Children with GI alterations and their families have many special needs. Some GI problems begin at birth, with life-threatening consequences. Some require the parents to accept their child’s altered appearance. Other problems develop after birth and provide long-term challenges in management and treatment. Sudden, unexpected surgery may be necessary. GI alterations cause anxiety and affect nutrition, elimination, respiratory status, skin integrity, body image, family processes, growth and development, and educational needs.
Upper and lower GI conditions can be categorized as follows:
• Obstructive disorders, such as pyloric stenosis, intussusception, and Hirschsprung disease
• Malabsorption conditions, such as lactose intolerance and celiac disease
• Hepatic disorders, such as hepatitis, biliary atresia, and cirrhosis
Disorders that involve the liver and biliary tract may be the result of congenital malformations or acquired infection. Because the liver is important to metabolism, alterations in its function can affect many body systems, including the cardiovascular, integumentary, renal, neurologic, hematologic, and immunologic systems. These disorders can also have significant effects on growth and development. Nursing care may involve nutritional support, infection control, developmental stimulation, family support, and intensive physiologic care during a period of crisis.
Disorders of Prenatal Development
Cleft Lip and Palate
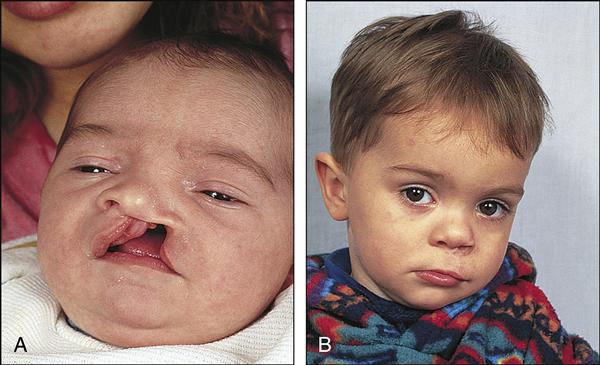
Cleft lip, cleft palate, and cleft lip and palate are separate anomalies that are closely related in etiology, pathophysiology, and nursing care. These distinct problems are all abnormal openings in the lip or palate. The defects may occur unilaterally (on either side) or bilaterally and are the most common congenital craniofacial deformity.
Incidence
The incidence ranges from 1 in 700 to 1000 births for cleft lip and palate and 1 in 2000 for cleft palate alone (Cleft Palate Foundation, 2007; March of Dimes, 2007; Zarate, Martin, Hopkin, et al., 2010). Cleft lip is seen predominantly in male infants and cleft palate in female infants. The prevalence of cleft lip and/or palate is higher in Asians and Native Americans and has a lower frequency in African-Americans. The etiology of cleft lip and palate malformations is thought to be multifactorial, including both genetic and environmental factors (Rojas-Martinez, Reutter, Chacon-Camacho, et al., 2010). While there appears
PATHOPHYSIOLOGY
Cleft Lip and Palate
Cleft lip and cleft palate occur from embryonic developmental failures related to multiple genetic and environmental factors. These developmental failures result in an abnormal opening in the lip, palate, and, sometimes, nasal cavity. Cleft lip results when the medial nasal and maxillary processes fail to join at 6 to 8 weeks of gestation. Cleft palate results from failure of the primary palatal shelves, or processes, to fuse at 7 to 12 weeks of gestation.
Each of these abnormalities appears as a distinct malformation, but they may also appear together. Achieving suction during feedings may be impossible, and fluids may enter the nose, putting the child at risk for aspiration, feeding difficulties, and respiratory distress.
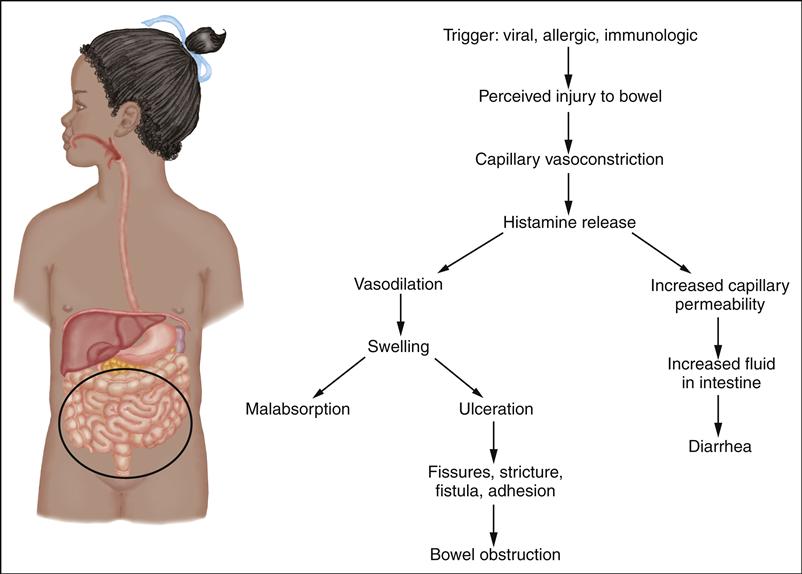
to be a genetic pattern or familial risk involved, environmental factors, such as maternal smoking, have been found to be associated with an increased risk of oral cleft defects (Lebby, Tan, & Brown, 2010). Many infants who are affected with cleft lip and palate have other associated defects.
Manifestations and Diagnostic Evaluation
Cleft lip has the following manifestations: a notched vermilion border, variably sized clefts that involve the alveolar ridge, and dental anomalies (usually deformed, supernumerary, or absent teeth). Cleft palate includes nasal distortion, midline or bilateral cleft with variable extension from the uvula and soft and hard palates, and exposed nasal cavities.
The diagnosis of cleft lip and cleft palate is based on observation at birth and complete examination in the neonatal period. Diagnosis may also be made in utero with ultrasound. Cleft lip is readily diagnosed through inspection of the lip. The first sign of cleft palate may be formula coming from the nose. A gloved finger placed in the mouth to feel the defect of the palate or visual examination with a flashlight confirms the diagnosis.
Therapeutic Management
Management is based on the severity of the defect. A number of professionals are involved in this process, including surgeons; nurses; geneticists; psychologists or psychiatrists; ear, nose, and throat specialists; audiologists; and occupational and speech therapists. Orthodontists and plastic surgeons become involved in the lengthy management. Pediatricians provide ongoing child health care.
The first intervention involves modifying feeding techniques as needed to allow adequate growth. Use of special feeding techniques, obturators, and unique nipples and feeders can usually accomplish this goal and allow early discharge home with parents (Figure 43-1). These modified techniques can decrease the energy required for the infant to take in adequate nutrition. Before surgical repair, removable orthopedic devices such as a Latham device may be used to expand and realign parts of the palate or decrease the size of a wide lip cleft.
Cleft lip repair is usually performed by age 3 to 6 months. Early repair may improve bonding and makes feeding much easier. The surgical technique involves the use of a staggered suture line to minimize scarring. Some cosmetic modifications may be needed again at age 4 to 5 years.
Cleft palate repair is individualized and based on the degree of deformity and size of the child. Closure is completed between ages 6 and 24 months. Most teams recommend repair by 1 year of age. Earlier closure facilitates speech development.
Concurrent treatment of altered dentition, recurring otitis media, speech dysfunction, emotional issues, and cosmetic concerns completes the ongoing therapy. Children with cleft palate are at high risk for developing chronic otitis media, which can cause long-term hearing loss.
Esophageal Atresia with Tracheoesophageal Fistula
Esophageal atresia (EA) and tracheoesophageal fistula (TEF) are congenital malformations in which the esophagus terminates before it reaches the stomach and/or a fistula is present that forms an unnatural connection between the esophagus and the trachea. Figure 43-2 portrays the most common type of EA with a distal TEF.
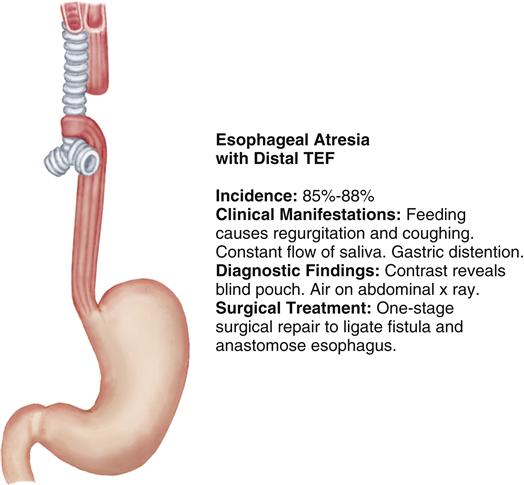
Etiology and Incidence
The cause of TEF and EA is unknown. EA with or without TEF occurs in 1.2 to 4.67 in 10,000 live births, with no difference by gender (National Birth Defects Prevention Network, 2009). Nearly half of infants born with EA have other associated anomalies of the cardiac, GI, and central nervous systems. Prematurity and low birth weight are frequent concomitant problems that significantly impact long-term prognosis.
Manifestations
Diagnostic Evaluation
A history of maternal polyhydramnios is a significant prenatal clue. If TEF is suspected prenatally, diagnosis can be made at the ideal time—in the delivery room. Atresia should be suspected if an NG tube cannot be passed 10 to 11 cm beyond the gum line. This suspicion is confirmed with an abdominal radiograph that
will identify a proximal esophagus dilated with air (atresia) or abdominal distention (fistula). The radiologist can identify the specific type of defect after instilling less than 1 mL of a water-soluble contrast medium into the NG tube and documenting its movement into the tracheal tree and the proximal pouch. This is then withdrawn from the pouch to minimize the risk of aspiration. Bronchoscopy and endoscopy are also used to identify and assess fistulas. The infant needs to have diagnostic testing for commonly associated cardiac and other congenital anomalies because these often are seen with this type of anomaly (Kahn & Orenstein, 2011a).
 NURSING CARE PLAN
NURSING CARE PLAN
The Child with a Cleft Lip or Palate
Focused Assessment
• During examination of the newborn with cleft lip and/or cleft palate, assess and document:
• Infant’s ability to suck, swallow, and breathe without distress
• Infant’s ability to handle normal secretions
• Parent’s initial reactions to the infant’s appearance
• Parent’s interactions with the infant (i.e., touching, holding, examining)
Nursing Diagnoses
Planning
Expected Outcomes
The child will:
Interventions and Rationales
1. Describe the degree of cleft lip and/or palate and impairment of sucking.
Infants with cleft lip alone or simple cleft dental arch may be successful with breastfeeding or bottle feeding without modifications.
2. Keep care and teaching simple and as closely related to normal infant feeding as possible.
Nutrition, parent-infant relationship, and adherence may be improved if normal techniques can be used.
3. Provide alternative assistive feeding devices as needed and ordered.
Some infants may be able to breastfeed successfully. Techniques and equipment vary among institutions. Use what is available and effective for each child. Encourage breastfeeding first because breastfeeding confers added immune protection.
4. Burp the infant frequently, and hold the infant in a more upright position.
Burping minimizes air swallowing and gastrointestinal (GI) flatus and minimizes risk of aspiration.
5. Document the feeding program in written form for parents to use at home, and provide the plan to other health professionals.
Documentation provides consistency at home and at other times when the family is in contact with numerous medical professionals treating the child.
6. Provide emotional support and positive reinforcement to parents as they learn to feed their child.
Self-care and bonding are improved when parents can assume total care.
7. Keep an accurate record of the child’s growth by using a growth chart.
A chart identifies growth changes early, when intervention can be most effective.
8. Explain preoperative and postoperative procedures: oral feedings withheld for 6 hours, placement of intravenous (IV) lines, use of elbow restraints, appearance of repair in the immediate postoperative period.
Explanation decreases parental anxiety and encourages involvement.
a. Keep straws, pacifiers, spoons, or fingers away from the child’s mouth for 7 to 10 days. Do not take temperatures orally.
Avoiding contact with the incision site reduces stress on surgical repair and prevents accidental tearing of very fine sutures.
b. Advance the child’s diet as ordered and tolerated from clear liquids to a normal soft diet within 48 hours.
A normal diet minimizes nutritional deficits and stress on the child. No foods that can tear surfaces are offered.
c. After repair of a cleft lip, resume preoperative feeding techniques.
Little evidence shows that sucking causes excess suture stress.
10. After repair of a cleft palate, provide short nipples that do not rest on palatal sutures; give baby food or baby food mixed with water.
Prevents direct contact with surgical site.
Evaluation
Nursing Diagnosis
Planning
Expected Outcomes
The parents will:
Interventions and Rationales
1. Encourage parents to discuss their fears, concerns, and negative emotions.
Grief, anxiety, confusion, guilt, denial, and anger are not uncommon and should be expressed.
2. Encourage touching and holding.
Contact encourages bonding and prevents a delayed attachment.
3. Make appropriate referral to a cleft lip and palate team of nurses, physicians, and other specialists as soon as possible.
A health care team can provide accurate information and begin to outline a plan of action.
4. Express acceptance of the baby by modeling feeding and close physical contact.
These interventions assist parents with the adaptation process.
5. Refer parents to community resources and parent groups.
Sharing with others in similar situations facilitates acceptance and adaptation.
6. Encourage parents to share concerns about long-term care and emotional and financial stress.
Long-term concerns require extensive follow-up and can strain many families’ resources. Identifying concerns early can increase problem-solving options.
Evaluation
Nursing Diagnoses
Planning
Expected Outcomes
Interventions and Rationales
1. Clean the lip repair site according to physician protocol. Many physicians recommend cleaning with sterile water by using a cotton swab or saline after feeding and as ordered. Use a rolling motion vertically down the suture line. Have parents demonstrate this cleaning technique.
The procedure decreases the medium for bacterial growth, decreases crusting, and minimizes scarring.
2. Apply antibiotic ointment as ordered.
Antibiotic ointment prevents infection, crusting, and scarring.
3. Use elbow restraints (no-no’s) to keep the child from touching the repair site. Continue for 6 to 8 days. Remove every 2 hours for 10 to 15 minutes. Remove restraint from only one elbow at a time, with a parent or nurse in constant attendance.
Elbow restraints prevent accidental rupture or tear of sutures. Periodically removing restraints promotes contact with the child, decreases anxiety, and allows the nurse to assess skin integrity and circulation.
4. Do not brush the child’s teeth for 1 to 2 weeks.
Avoiding brushing prevents accidental tear of palatal sutures.
5. Keep the child in a supine position or in an infant seat.
Careful positioning prevents contact of suture lines with bed linens.
6. Observe for redness, swelling, excessive bleeding, drainage, respiratory distress, or fever.
Signs of infection must be identified early because additional inflammation can increase scarring.
7. To clean the palate repair site, rinse the child’s mouth with water after feedings.
Rinsing after feeding removes food and residual sugars from suture lines, reducing the risk of infection.
8. Encourage the parents to hold and cuddle the child as the child desires.
Crying puts additional stress on the suture line.
9. Maintain lip protective devices if ordered.
Protective devices prevent separation of lip suture lines.
Evaluation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


