Tumors can impact the spinal cord by causing external compression or by directly involving the cord or intradural space. Metastatic lesions causing external compression are by far the most common.1 Primary tumors of the spinal cord constitute approximately 0.5% of newly diagnosed tumors and 7% of primary central nervous system neoplasms.1 The exact incidence of metastatic disease affecting the spinal cord is not known, but is thought to impact up to 20% of all cancer patients. In general, the underlying etiology is not known. The incidence and types of spinal cord tumors vary greatly between children and adults. Only tumors affecting adults are discussed in this chapter.
CLASSIFICATION
As with tumors involving the brain, spinal cord tumors have several classification schemas. The histology classification of spinal cord tumors includes primary tumors, which arise from the constituent elements of the central nervous system, or metastatic tumors, which spread from tumors originating in other organs. Table 21-1 provides the common histologic classification of tumors. This system provides information on the histologic origin of the tumor as well as the grade, or degree of malignancy. From a histologic perspective, most primary spinal cord tumors are benign. Spinal cord tumors are also classified according to their anatomic location with reference to the dural meningeal covering and the spinal cord, or to the location along the vertebral column.
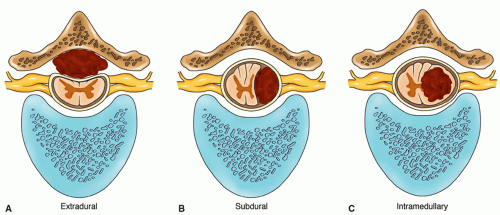
Location in Relation to Dura and Spinal Cord. Spinal cord tumors may be classified as extramedullary or intramedullary with respect to the dura and spinal cord.
Extramedullary tumors are located outside the spinal cord and are the most common location for primary spinal cord tumors, accounting for about 80% to 90% of cases.2
Extramedullary tumors are subdivided into the following.
Extradural are located outside the spinal dura; within the epidural space. Extradural tumors most often involve the vertebrae and may also contain a soft tissue component. The most common primary tumors are chordomas and sarcomas— representing 20% of all primary spinal cord tumors. This is also the most common location for metastatic cancer, with the most common primary sites being lung, breast, prostate, colon, kidney, and uterus. While less common, hematopoietic malignancies such as myeloma or lymphoma can also occupy the epidural space.3
Intradural are located within the spinal dura, but not within the spinal cord. Primary intradural tumors include meningiomas, neurofibromas, and schwannomas, which constitute 60% of all primary spinal cord tumors.4
Meningiomas can grow adjacent to the nerve root without direct involvement of the nerve root whereas neurofibromas and schwannomas arise from the peripheral nerves and occur in association with the nerve root often extending through the neural foramen. Schwannomas most commonly occur as single lesions and are confined to the nerve sheath. In contrast, neurofibromas invade the nerve, and axons can be found between the tumor cells. Clinical evidence of multiple neurofibromas or schwannomas are suggestive of genetic disorders which are characterized by multiple tumors throughout the nervous system. Neurofibromatosis Type 1 gene (NF1) is a fairly common genetic disease occurring in 1:3000 individuals. While sporadic mutations occur, 50% of cases with multiple neurofibromas are due to genetic transmission.
Note that 2% to 4.6% of NF 1 gene carriers are predisposed to a malignant transformation of neurofibroma known as malignant peripheral nerve sheath tumor (MPNST). MPNST is thought to develop as a result of additional mutations involving tumor-suppressor genes. The Neurofibromatosis Type 2 gene (NF2) is a less common genetic disease occurring in 1:40,000 and is associated with multiple schwannomas or bilateral acoustic neuromas. While patients with NF2 can develop tumors throughout the body, almost all individuals with NF2 will develop bilateral tumors involving the eighth cranial nerve. Both primary and metastatic cancer can also involve the cerebrospinal fluid, often called leptomeningeal disease (LMD). Approximately 16,000 patients are diagnosed each year with LMD. The most common tumors to cause LMD are breast and lung cancer and lymphoma.5
Intramedullary tumors are located within the substance of the spinal cord.
Intramedullary tumors account for approximately 5% to 10% of all spinal tumors, with ependymomas and gliomas being the most common. Hemangioblastoma, the third most common intramedullary tumor, accounts for only 3% to 8% of all intramedullary spinal cord tumors, and can be associated with Von Hippel-Lindau Syndrome, a genetic disorder characterized by the development of malignant or benign tumors.1 A subtype of non-Hodgkin’s lymphoma, primary central nervous system lymphoma (PCNSL) can affect the spinal cord and other segments of the neuroaxis. Immunodeficiency is the only known risk factor for developing PCNSL.6 Metastasis from a systemic cancer to the intramedullary space is exceptionally rare. The locations of primary spinal cord tumors are shown on cross section in Figure 21-1.
TABLE 21-1 COMMON SPINAL CORD TUMORS
TUMOR TYPE
HIGHEST INCIDENCE
GENDER
DESCRIPTION
Neurofibroma
30-50 yrs
Equally affected
60% above L-1, causing spastic paraparesis; 30% below L-1, causing a lateral cauda equina syndrome. Multiple tumor growths and tumor involving the bilateral eight cranial nerve (Acoustic Neuromas) are commonly associated with NF2 abnormalities)
Meningioma
M/F ratio 1:9
Most in midthoracic (T-3-6); a few in foramen magnum
Ependymomas
Average age 30 yrs
M/F ratio 2:1
Either intrinsic within the spinal cord at C-6-T-2 or on the filum terminate (central cauda equina syndrome). Very high incidence of associated syringomyelic cavitation, making the lesion seem much more extensive than its actual size
Gliomas
35-45 yrs
Equally affected
Almost all in cervical cord
Dermoids
15-20 yrs
No data
Located in sacrococcygeal area, usually associated with spina bifida
Chordoma
40-70 yrs
Mostly males
Most in sacrococcygeal area; bone destruction and pain are major feature
Hemangioblastoma
Before the age of 40
No data
Well-circumscribed, encapsulated vascular tumor that can usually be completely removed; accounts for 3-8% of intramedullary tumors
Schwannomas
30-50 yrs
Equally affected
70-75% intradural extramedullary. Radiographic solitary ‘dumbbell’ shaped mass (not associated with NF2 gene).
Clinical Pearl: Extradural tumors are the most common type of spinal cord tumor.
Intradural tumors—Multiple neurofibromas, schwannomas, and bilateral acoustic schwannomas are highly indicative of a genetic anomaly (NF1 or NF2 gene). Ependymoma is the most common intramedullary tumor.
Location in Relationship to the Vertebral Column. Spinal cord tumors are roughly distributed as follows: cervical, 30%; thoracic, 50%; and lumbosacral, 20%. Locating a tumor more precisely helps to correlate the dermatome with specific functional assessment for that level (see Chapters 5 and 18). Conversely, identifying deficits on examination can help to diagnose a spinal cord tumor and identify its location according to the involved dermatome.
Figure 21-1 ▪ Location of intramedullary and extramedullary (subdural and extradural) tumors on cross section.
Classification according to histopathologic tumor type: as mentioned previously, spinal cord tumors can be primary (arising from the spinal cord) or metastatic (spread from another primary site). Primary spinal cord tumors include neurofibromas, schwannomas, and meningiomas usually found in the extramedullary area, and ependymomas and other gliomas usually found in the intramedullary area. Other types of primary spinal cord tumors include sarcomas, vascular tumors, chordomas, and epidermoids (Table 21-1). Metastatic spinal cord tumors are mostly extradural tumors. Although malignancies from almost any site can metastasize to the vertebral column, lesions from the lungs, breast, prostate, colon, kidney, and uterus, as well as lymphomas and multiple myelomas, are the most common.
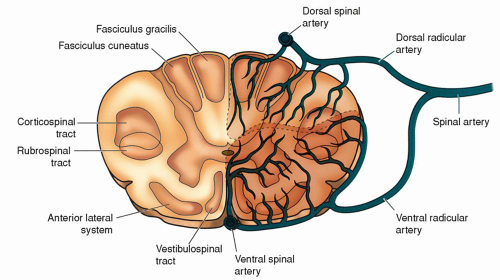
Figure 21-2 ▪ Perfusion patterns of the spinal cord. (From: Kingley, R. E. (2000) Concise text of neuroscience. Philadelphia: Lippincott Williams & Wilkins.)
PATHOPHYSIOLOGY
Regardless of the type or location of a spinal cord tumor, the associated pathophysiologic changes can lead to cord dysfunction and neurological deficits. These changes can result from direct cord compression, ischemia secondary to arterial or venous obstruction, or, in the case of intramedullary tumors, direct invasion (Figs. 21-2 and 21-3). Tumors causing cord compression can cause traction on or irritation of the spinal nerve roots, displacement of the spinal cord, interference with the spinal blood supply, or obstruction of cerebrospinal fluid (CSF) circulation. Cord compression alters the normal physiology involved in providing an adequate blood supply, maintaining stable cellular membranes, and facilitating afferent and efferent impulses for specific sensory, motor, and reflex functions of the spinal cord and related spinal nerves.
Invasive infiltration of the spinal cord is seen in association with intramedullary tumors. Edema associated with cord compression is seen in both extramedullary and intramedullary tumors. Edema can ascend the spinal cord, causing additional deficits to the sensitive cord. Control of edema is a major focus in management and will be discussed later. Focal (localizing) signs depend on the spinal level, the cross-section location, and the rate of growth and density of the spinal tumor.
Clinical Pearl: Pathophysiologic changes can result from direct cord compression, ischemia secondary to arterial or venous obstruction, or, in the case of intramedullary tumors, direct invasion.
Spinal Level
Spinal level refers to the specific location of the tumor in the cervical, thoracic, or lumbosacral area. Sensory distribution from the spinal nerves at each level is mapped at dermatome levels (see chapters 5 and 17). Loss of sensation depends on the level of the tumor and correlates with the dermatome level. Motor deficits are also related to the tumor level, because motor components of various spinal nerves come together to innervate specific areas.
Clinical Pearl: Beginning at T2, the thoracic nerve roots innervate the intercostal, abdominal, and paraspinal muscles; therefore, assessment of thoracic nerve root damage is most adequately evaluated by sensory exam. However, considerable overlap of sensory innervation between adjacent dermatomes can occur. As a result, loss of afferent nerve function at any level from T2 to T12, would not necessarily result in complete loss of sensation, but rather a decrease in sensation can be experienced.
Location on Cross Section
Tumors involving the vertebral elements often cause anterior cord compression, because metastatic tumors frequently involve the vertebral body. In the anterior location, a pathologic fracture of the vertebral body with bone fragments driven back into the cord or tumor growth can encroach on the anterior aspect of the spinal cord. Isolated posterior element involvement is much less common.
The major sensations conveyed by the spinal cord are light touch, pain and temperature, and position and vibration. A specific tract located in a designated anatomic area, noted on cross section (see chapter 5), carries each sensation. The posterior columns (fasciculus gracilis and fasciculus cuneatus) convey position and vibration sensations. The lateral spinothalamic tracts convey pain and temperature. The anterior spinothalamic tract conveys light touch. On cross section, the spinothalamic tracts are located in the anterolateral area of the spinal cord (Fig. 21-2). Therefore, the location of the tumor on cross section will help determine functional loss.
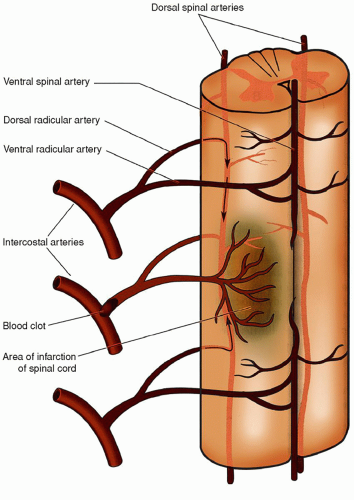
Figure 21-3 ▪ Radicular arteries of the spinal cord. Not all of the 31 potential pairs of spinal arteries develop completely. Therefore, some parts of the spinal cord are perfused by only one or two radicular spinal arteries. Loss of a single artery at these sites can result in ischemic infarction of the spinal cord. The upper lumbar and thoracic spinal cord are particularly vulnerable. (From: Kingley, R. E. (2000). Concise text of neuroscience. Philadelphia: Lippincott Williams & Wilkins.)
Rate of Growth and Density
The development of neurological deficits is directly related to the tumor’s rate of growth and its density (i.e., soft or hard). A slowgrowing tumor can accommodate itself to the limited space of the vertebral-spinal cord space within compensatory limits. Many primary spinal cord tumors grow very slowly over years, compressing the cord into a thin, ribbon-like structure with minimal neurological deficits. However, with a rapidly growing tumor, such as a primary malignant spinal cord tumor or a metastatic lesion, the cord fares poorly, and neurological deficits occur and can be permanent. Physiologically, the tumor produces major compression of the cord, resulting in substantial edema and, possibly, rapid paralysis (within hours).
A soft tumor can be of a consistency similar to that of the spinal cord. If it grows slowly, it causes gradual compression of the spinal cord. However, the cord’s blood supply is able to respond to this alteration and adequately supply the vascular needs without interruption. The tumor adjusts to the available space for growth, becoming elongated as necessary. Neither movement of the spinal column nor normal alterations in blood flow to the cord will produce injury by contusion or ischemia. By contrast, a hard tumor will respond to movement or vascular changes with spinal contusion, ischemia, and irreversible cord damage. Encroachment on neurological function is apparent earlier with hard tumors than with soft tumors because hard tumors do not conform to the available space.
Morphologically, spinal cord tumors are described as encapsulated or sharply outlined. Soft tumors are irregularly elongated and can extend for two or more segments whereas hard tumors are more circumscribed. Some tumors have a fluid-filled cystic cavity called a syrinx that may require drainage.
Metastatic Spinal Cord Tumors
Spinal cord compression from metastases occurs in about 20% of all cancer patients. An epidural spinal tumor is the first sign of a malignancy in about 10% of patients.7 Metastatic lesions can involve the vertebrae, grow through the intravertebral foramina, or directly impact the dura, spinal fluid, or spinal cord. Malignant cells spread to the spinal region and adjacent tissues via the venous system, arterial system, and the cerebrospinal fluid. The arterial system may transmit cancer cells to the bony structures of the spine, where tumor cells thrive in a bone marrow rich environment. In this favorable setting, the malignant cells multiply, causing bony destruction and subsequent compression of the spinal cord and nerves. The unique valveless venous system surrounding the spinal cord can also transmit cancer cells into the epidural space, where growth can result in compression within the dural sac.
Tumor cells can also travel by way of the cerebrospinal fluid and become entrapped among the roots of the cauda equina resulting in “drop metastasis.” The vertebral column is the most common site in the bony skeletal system for metastases. As mentioned previously, the most common primary sites of metastatic spinal cord tumors are the lungs, breast, prostate, colon, kidneys, and uterus. The malignant lesion tends to spread proximally from the primary organ affected to the adjacent vertebral body, spinous or transverse process, or pedicle. Progressive tumor within the vertebral body causes bony destruction or vertebral collapse with or without spinal instability. Evidence of metastatic involvement is not visible on plain x-rays until approximately 30% to 50% of the vertebral body is destroyed. In this scenario, plain x-rays may demonstrate decrease vertebral height or a ventral wedge compression producing a kyphotic deformity8. The lesion then causes epidural compression of the spinal cord or nerve roots. Tumors occur in the thoracic spine (70% of cases) followed by the lumbar spine (20%). Over 30% of patients have involvement at multiple spine levels. Retroperitoneal neoplasms (especially lymphomas or sarcomas) may enter the spinal canal through the intervertebral foramina.
Typically extradural tumors produce radicular pain and signs of root involvement prior to cord compression. Metastatic lesions most commonly exert their effects on the spinal cord by direct compression, enlargement of a vertebral body or pedicle, collapse of an eroded vertebra, or direct extension of a paravertebral lesion through the intervertebral foramen. Edema of the cord, a consequence of compression, increases the degree of the deficit.
SIGNS AND SYMPTOMS ASSOCIATED WITH SPINAL CORD TUMORS
Symptoms may develop insidiously and progress gradually, or they may progress rapidly, as is common with metastatic lesions. The specific signs and symptoms depend on the anatomic location of the tumor (location on cross section), its location in relation to the level of the vertebral column (thoracic, lumbosacral), and the specific spinal nerves involved. Focal signs and symptoms aid in determining the level of the lesion; however, they can also be indistinct, misleading, and intermittently variable. Table 21-2 summarizes signs and symptoms according to the anatomic level of the tumor on or within the spinal cord.
Pain is the initial symptom in almost all patients. About 95% of patients with vertebral or spinal cord tumors have pain without neurological deficits as the initial symptom. At the time of diagnosis, 75% have pain and weakness, sensory disturbance, or sphincter dysfunction.9 This highlights the importance of prompt diagnosis in order to preserve neurological function.
Clinical Pearl: The specific signs and symptoms depend on the anatomic location of the tumor (location on cross section), its location in relation to the level of the vertebral column (thoracic, lumbosacral), and the specific spinal nerves involved.
Pain
The pain may be aching and localized or sharp and radiating (radicular pain). Localized pain and point tenderness are common over the involved vertebral area when pressure is applied to the spinous process. Pain, which can localize to the back or to an extremity, is caused by irritation, tension/traction, or compression of the nerve root. The quality of pain can vary from mild to severe and from dull to piercing, but it is almost always present. Radicular pain results from compression of the dorsal nerve root, and is defined as pain within the sensory distribution of a spinal nerve root. It can be characterized by both numbness and pain. Compression of the dorsal nerve root irritates the neurons causing spontaneous firing of the nerve, resulting in a feeling of both pain and paresthesias which can occur with minimal stimulation. Radicular pain often has a neuropathic component which is commonly described as sharp and burning.10 Any activity that increases intraspinal pressure, such as the Valsalva maneuver (as occurs with coughing, sneezing, or straining) and movement, can cause pain to intensify and radiate. Pain can also be exaggerated by reclining, because this position stretches the spinal nerves. As a result, pain may awaken the patient at night.
Pain typically precedes signs of cord compression by weeks or even months, but after cord compression occurs, it is always progressive and may advance rapidly.7 Pain can be focal or radiate in a known dermatomal pattern. Because the dermatomes supplying a particular area of the body overlap, pain can also be diffuse, mimicking such conditions as angina, an acute abdominal lesion, or intercostal neuralgia. Focal pain may be localized to the level of the lesion and is often associated with expansion of the vertebral elements by tumor or vertebral fracture. Focal pain may worsen upon standing (load-bearing pain) or with movement (mechanical pain).
Clinical Pearl: The most common presenting symptom of spinal cord tumor is pain.
Motor Deficits
The presenting motor signs and symptoms depend on the degree of involvement of the spinal nerve root and the spinal cord (Fig. 21-4). For example, involvement of the anterior spinal nerve root leads to a lower motor neuron syndrome. This is characterized by motor weakness, wasting and fasciculations of involved muscles, hypotonia (flaccidity), loss of tendon reflexes when neurons responsible for those reflexes are affected, and normal abdominal and plantar reflexes unless the neurons responsible for those reflexes are directly involved, in which case the reflex response is lost.
When the spinal cord becomes compressed, other motor deficits develop. Involvement of motor tracts results in an upper motor neuron deficit below the level of the lesion. An upper motor neuron syndrome includes weakness or paralysis, spasticity, increased tendon reflexes, a positive Babinski sign, loss of abdominal reflexes, and little or no muscle atrophy.
The patient can have a mixed presentation of lower motor neuron disease and upper motor neuron disease, depending on the degree of compression present and the anatomic structures affected. Additionally, a combination of sensory and motor deficits may be noted as a Brown-Séquard syndrome (loss of motor function, light touch, vibration, and position sense on the side of the lesion, and contralateral loss of pain and temperature sense).
Sensory Deficits
Which specific sensory deficits develop depends on the presentation of the tumor on cross section. A lateral presentation affects pain and temperature sensation. Numbness or paresthesias, especially of the legs, appear as early symptoms. Light touch sensation is preserved in the presence of a unilateral tumor resulting from the crossed and uncrossed components to the tract. Awareness of vibration and proprioception of body parts are affected if the posterior columns are involved. Compression from a tumor affects function below the lesion. Therefore, the highest intact sensory level should be determined. An intramedullary lesion may result in a “suspended sensory level” in which a band-like loss of sensation occurs at the level of the lesion.
TABLE 21-2 SIGNS AND SYMPTOMS OF SPINAL CORD TUMORS BY VERTEBRAL LEVEL
LOCATION
SIGNS AND SYMPTOMS
COMMENTS
Cervical Levels
C-4 and above
Especially dangerous because of innervation to the diaphragm (C-1-4) and the potential effect on respirations
High cervical tumors can affect the lower cranial nerves (VIII to XII).
Possible respiratory difficulty
Quadriparesis or quadriplegia
Paresthesia
Occipital headache
Stiff neck
CN VIII: downbeat nystagmus
CNs IX and X: dysphagia; dysarthria
CN XI: difficulty shrugging shoulders; atrophy of shoulder and neck muscles
CN XII: deviation of tongue; difficulty speaking; unilateral tongue atrophy
Difficult surgical access; may consider proton beam therapy or another nonsurgical modality
Downward gaze is controlled by pathways that extend from the brainstem to the upper cervical cord.
Below C-4
Pain in shoulders and arms
Paresthesia
If the C-5-6 root is involved, there will be pain along the medial aspect of the arm.
If C-7-8 is involved, there will be pain along the outer side of the forearm and hand.
Weakness follows pain.
Atrophy of the shoulder, arm, and intrinsic hand muscles is often associated with fasciculation.
Horner’s syndrome (ptosis, miosis, and anhidrosis on the affected side)
Hyperactive reflexes
Attributable to interference with sympathetic innervation
Thoracic Levels
T-1-12
Most metastatic lesions involve the thoracic region.
Pain in chest/back
Use of motor deficits to localize the lesion is difficult; spastic paresis may be evident.
Sensory deficits are more accurate in identifying the lesion’s level:
– Know landmark areas, such as T-4 (nipple line) and T-10 (umbilicus).
– A band of hyperesthesia is often found above the level of the lesion.
A positive Babinski sign is noted.
Bowel and/or bladder dysfunction
Sexual dysfunction
See Chapter 18 for an explanation and illustration of dermatomes.
Lumbosacral
S-1-5
Pain in lower back, which often radiates to legs; may also be felt in the perineal area
Paresis/spasticity of lower extremities, usually in one leg and later in the other
Sensory loss in legs and/or saddle area
Bowel and/or bladder dysfunction
Sexual dysfunction
Reflexes—ankle and knee-jerk reflexes are diminished or absent
Footdrop is common.
Atrophy may affect certain muscle groups.
CN, cranial nerve.
Only gold members can continue reading. Log In or Register to continue