CHAPTER 4 There are four basic pharmacokinetic processes: absorption, distribution, metabolism, and excretion (Fig. 4–1). Absorption is defined as the movement of a drug from its site of administration into the blood. Distribution is defined as drug movement from the blood to the interstitial space of tissues and from there into cells. Metabolism (biotransformation) is defined as enzymatically mediated alteration of drug structure. Excretion is the movement of drugs and their metabolites out of the body. The combination of metabolism plus excretion is called elimination. The four pharmacokinetic processes, acting in concert, determine the concentration of a drug at its sites of action. As a nurse, you will have ample opportunity to apply knowledge of pharmacokinetics in clinical practice. For example, by understanding the reasons behind selection of route, dosage, and dosing schedule, you will be less likely to commit medication errors than will the nurse who, through lack of this knowledge, administers medications by blindly following prescribers’ orders. Also, as noted in Chapter 2, prescribers do make mistakes. Accordingly, you will have occasion to question or even challenge prescribers regarding their selection of dosage, route, or schedule of administration. In order to alter a prescriber’s decision, you will need a rational argument to support your position. To present that argument, you will need to understand pharmacokinetics. The basic structure of the cell membrane is depicted in Figure 4–2. As indicated, the basic membrane structure consists of a double layer of molecules known as phospholipids. Phospholipids are simply lipids (fats) that contain an atom of phosphate. In Figure 4–2, the phospholipid molecules are depicted as having a round head (the phosphate-containing component) and two tails (long-chain hydrocarbons). The large objects embedded in the membrane represent protein molecules, which serve a variety of functions. Polar molecules are molecules with uneven distribution of electrical charge. That is, positive and negative charges within the molecule tend to congregate separately from one another. Water is the classic example. As depicted in Figure 4–3A, the electrons (negative charges) in the water molecule spend more time in the vicinity of the oxygen atom than in the vicinity of the two hydrogen atoms. As a result, the area around the oxygen atom tends to be negatively charged, whereas the area around the hydrogen atoms tends to be positively charged. Kanamycin (Fig. 4–3B), an antibiotic, is an example of a polar drug. The hydroxyl groups, which attract electrons, give kanamycin its polar nature. Quaternary ammonium compounds are molecules that contain at least one atom of nitrogen and carry a positive charge at all times. The constant charge on these compounds results from atypical bonding to the nitrogen. In most nitrogen-containing compounds, the nitrogen atom bears only three chemical bonds. In contrast, the nitrogen atoms of quaternary ammonium compounds have four chemical bonds (Fig. 4–4A). Because of the fourth bond, quaternary ammonium compounds always carry a positive charge. And because of the charge, these compounds are unable to cross most membranes. Tubocurarine (Fig. 4–4B) is a representative quaternary ammonium compound. Until recently, purified tubocurarine was employed as a muscle relaxant for surgery and other procedures. A crude preparation—curare—is used by South American Indians as an arrow poison. When employed for hunting, tubocurarine (curare) produces paralysis of the diaphragm and other skeletal muscles, causing death by asphyxiation. Interestingly, even though meat from animals killed with curare is laden with poison, it can be eaten with no ill effect. Why? Because tubocurarine, being a quaternary ammonium compound, cannot cross membranes, and therefore cannot be absorbed from the intestine; as long as it remains in the lumen of the intestine, curare can do no harm. As you might gather, when tubocurarine was used clinically, it could not be administered by mouth. Instead, it had to be injected. Once in the bloodstream, tubocurarine then had ready access to its sites of action on the surface of muscles. A review of acid-base chemistry should help. An acid is defined as a compound that can give up a hydrogen ion (proton). Put another way, an acid is a proton donor. A base is defined as a compound that can take on a hydrogen ion. That is, a base is a proton acceptor. When an acid gives up its proton, which is positively charged, the acid itself becomes negatively charged. Conversely, when a base accepts a proton, the base becomes positively charged. These reactions are depicted in Figure 4–5, which shows aspirin as a representative acid and amphetamine as a representative base. Because the process of an acid giving up a proton or a base accepting a proton converts the acid or base into a charged particle (ion), the process for either an acid or a base is termed ionization. The process whereby a drug accumulates on the side of a membrane where the pH most favors its ionization is referred to as ion trapping or pH partitioning. Figure 4–6 shows the steps of ion trapping using aspirin as an example. For each of the major routes of administration—oral (PO), intravenous (IV), intramuscular (IM), and subcutaneous (subQ)—the pattern of drug absorption (ie, the rate and extent of absorption) is unique. Consequently, the route by which a drug is administered will significantly affect both the onset and the intensity of effects. Why do patterns of absorption differ between routes? Because the barriers to absorption associated with each route are different. In the discussion below, we examine these barriers and their influence on absorption pattern. In addition, as we discuss each major route, we will consider its clinical advantages and disadvantages. The distinguishing characteristics of the four major routes are summarized in Table 4–1. TABLE 4–1 Properties of Major Routes of Drug Administration
Pharmacokinetics
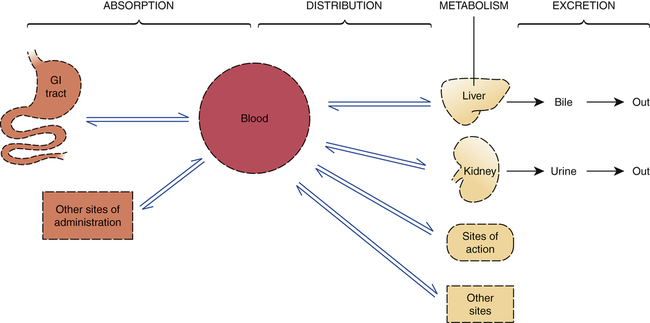
 The four basic pharmacokinetic processes.
The four basic pharmacokinetic processes.
Dotted lines represent membranes that must be crossed as drugs move throughout the body.
Application of pharmacokinetics in therapeutics
Passage of drugs across membranes
Membrane structure
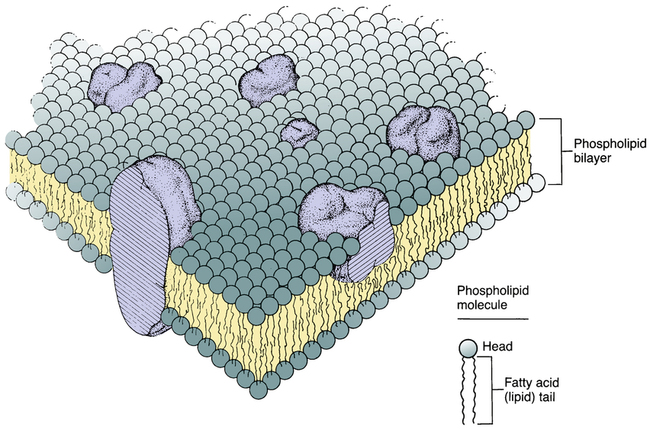
 Structure of the cell membrane.
Structure of the cell membrane.
The cell membrane consists primarily of a double layer of phospholipid molecules. The large globular structures represent protein molecules imbedded in the lipid bilayer. (Modified from Singer SJ, Nicolson GL: The fluid mosaic model of the structure of cell membranes. Science 175:720, 1972.)
Polar molecules
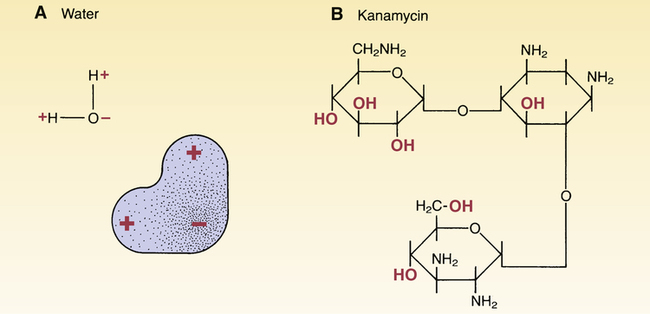
 Polar molecules.
Polar molecules.
A, Stippling shows the distribution of electrons within the water molecule. As indicated, water’s electrons spend more time near the oxygen atom than near the hydrogen atoms, making the area near the oxygen atom somewhat negative and the area near the hydrogen atoms more positive. B, Kanamycin is a polar drug. The 2 –OH groups of kanamycin attract electrons, thereby causing the area around these groups to be more negative than the rest of the molecule.
Ions
Quaternary ammonium compounds
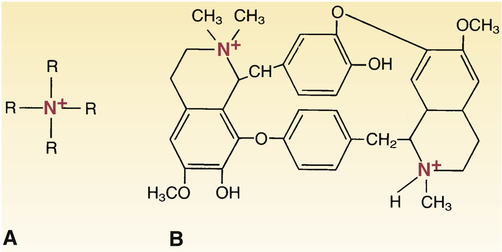
 Quaternary ammonium compounds.
Quaternary ammonium compounds.
A, The basic structure of quaternary ammonium compounds. Because the nitrogen atom has bonds to four organic radicals, quaternary ammonium compounds always carry a positive charge. Because of this charge, quaternary ammonium compounds are not lipid soluble and cannot cross most membranes. B, Tubocurarine is a representative quaternary ammonium compound. Note that tubocurarine contains two “quaternized” nitrogen atoms.
PH-dependent ionization
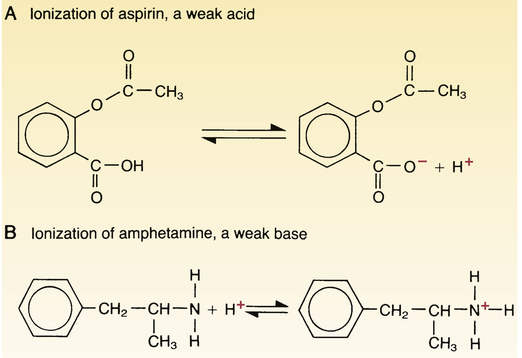
 Ionization of weak acids and weak bases.
Ionization of weak acids and weak bases.
The extent of ionization of weak acids (A) and weak bases (B) depends on the pH of their surroundings. The ionized (charged) forms of acids and bases are not lipid soluble and hence do not readily cross membranes. Note that acids ionize by giving up a proton and that bases ionize by taking on a proton.
Ion trapping (pH partitioning)
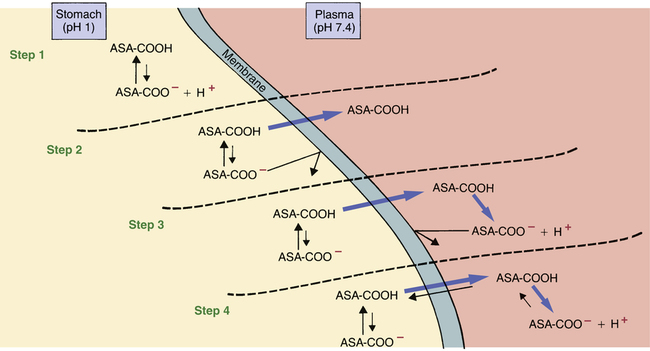
 Ion trapping of drugs.
Ion trapping of drugs.
This figure demonstrates ion trapping using aspirin as an example. Because aspirin is an acidic drug, it will be nonionized in acid media and ionized in alkaline media. As indicated, ion trapping causes molecules of orally administered aspirin to move from the acidic (pH 1) environment of the stomach to the more alkaline (pH 7.4) environment of the plasma, thereby causing aspirin to accumulate in the blood. In the figure, aspirin (acetylsalicylic acid) is depicted as ASA with its COOH (carboxylic acid) group attached.
Step 1: Once ingested, ASA dissolves in the stomach contents, after which some ASA molecules give up a proton and become ionized. However, most of the ASA in the stomach remains nonionized. Why? Because the stomach is acidic, and acidic drugs don’t ionize in acidic media.
Step 2: Because most ASA molecules in the stomach are nonionized (and therefore lipid soluble), most ASA molecules in the stomach can readily cross the membranes that separate the stomach lumen from the plasma. Because of the concentration gradient that exists between the stomach and the plasma, nonionized ASA molecules will begin moving into the plasma. (Note that, because of their charge, ionized ASA molecules cannot leave the stomach.)
Step 3: As the nonionized ASA molecules enter the relatively alkaline environment of the plasma, most give up a proton (H+) and become negatively charged ions. ASA molecules that become ionized in the plasma cannot diffuse back into the stomach.
Step 4: As the nonionized ASA molecules in the plasma become ionized, more nonionized molecules will pass from the stomach to the plasma to replace them. This movement occurs because the laws of diffusion demand equal concentrations of diffusible substances on both sides of a membrane. Because only the nonionized form of ASA is able to diffuse across the membrane, it is this form that the laws of diffusion will attempt to equilibrate. Nonionized ASA will continue to move from the stomach to the plasma until the amount of ionized ASA in plasma has become large enough to prevent conversion of newly arrived nonionized molecules into the ionized form. Equilibrium will then be established between the plasma and the stomach. At equilibrium, there will be equal amounts of nonionized ASA in the stomach and plasma. However, on the plasma side, the amount of ionized ASA will be much larger than on the stomach side. Because there are equal concentrations of nonionized ASA on both sides of the membrane but a much higher concentration of ionized ASA in the plasma, the total concentration of ASA in plasma will be much higher than in the stomach.
Absorption
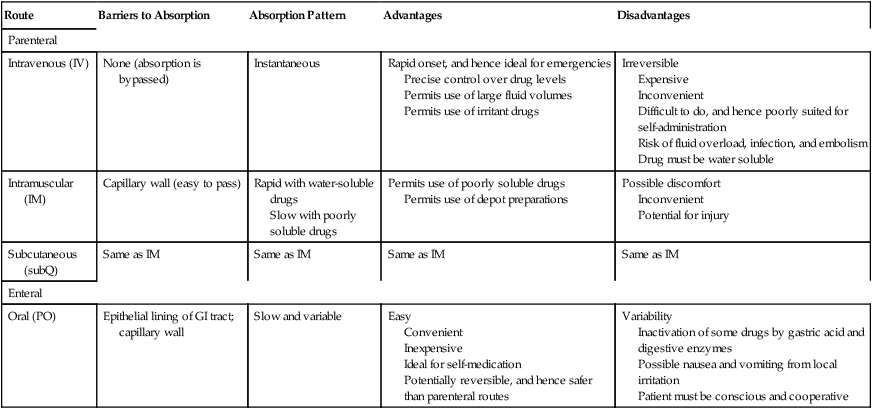
Characteristics of commonly used routes of administration

Route
Barriers to Absorption
Absorption Pattern
Advantages
Disadvantages
Parenteral
Intravenous (IV)
None (absorption is bypassed)
Instantaneous
Rapid onset, and hence ideal for emergencies
Precise control over drug levels
Permits use of large fluid volumes
Permits use of irritant drugs
Irreversible
Expensive
Inconvenient
Difficult to do, and hence poorly suited for self-administration
Risk of fluid overload, infection, and embolism
Drug must be water soluble
Intramuscular (IM)
Capillary wall (easy to pass)
Rapid with water-soluble drugs
Slow with poorly soluble drugs
Permits use of poorly soluble drugs
Permits use of depot preparations
Possible discomfort
Inconvenient
Potential for injury
Subcutaneous (subQ)
Same as IM
Same as IM
Same as IM
Same as IM
Enteral
Oral (PO)
Epithelial lining of GI tract; capillary wall
Slow and variable
Easy
Convenient
Inexpensive
Ideal for self-medication
Potentially reversible, and hence safer than parenteral routes
Variability
Inactivation of some drugs by gastric acid and digestive enzymes
Possible nausea and vomiting from local irritation
Patient must be conscious and cooperative
Intravenous
Disadvantages.
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


