(PEN-toh-stah-tin)
2-Deoxycoformycin, Nipent
Antineoplastic (antibiotic)
pH 7 to 8.5
Usual dose
Evaluation and prehydration are required before administration; see Monitor.
4 mg/M2 every other week. Do not exceed recommended dose; see Precautions. If there is no major toxicity and improvement is continuous, treat until a complete response is achieved, then administer two additional doses. Do not treat beyond 12 months.
Dose adjustments
Reduced dose and benefit-versus-risk assessment may be required with impaired renal function (CrCl below 60 mL/min); insufficient data available. Two patients with impaired renal function (CrCl 50 to 60 mL/min) achieved complete response without unusual adverse events when treated with 2 mg/M2. ■ Withhold dose if SCr elevated; obtain CrCl. ■ Withhold dose if the absolute neutrophil count falls from a baseline of greater than 500 cells/mm3 before therapy to less than 200 cells/mm3 during treatment. Resume treatment when count returns to predose levels.
Dilution
Specific techniques required; see Precautions. Diluent (5 mL SWFI) provided; dissolve completely; will yield 2 mg/mL. May be given by IV injection or further diluted in 25 to 50 mL NS or D5W; 25 mL yields 0.33 mg/mL, 50 mL yields 0.18 mg/mL. Treat spills or waste with a 5% sodium hypochlorite solution before disposal.
Storage:
Refrigerate before initial reconstitution. Store at room temperature and use within 8 hours after initial reconstitution or dilution for infusion.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Does not interact with PVC infusion containers or administration sets at concentrations specified for dilution.
One source suggests the following compatibilities:
Y-site:
Melphalan (Alkeran), ondansetron (Zofran), paclitaxel (Taxol), sargramostim (Leukine). Physically compatible at the Y-site with fludarabine (Fludara); however, the two drugs are not recommended for concurrent use; see Drug/Lab Interactions.
Rate of administration
Follow each dose with an additional 500 mL of prehydration infusion fluids.
IV injection:
A single dose over 1 minute.
Infusion:
A single dose over 20 to 30 minutes.
Actions
Mechanism of action is not known, but it is cytotoxic as a result of its potent inhibition of the enzyme adenosine deaminase (ADA). Blocks DNA and RNA synthesis and causes DNA damage. Average terminal half-life of 6 hours is extended to 18 hours in patients with impaired renal function (CrCl less than 50 mL/min). Inhibits ADA for up to 1 week; actual response may not occur for months. Primarily excreted in urine.
Indications and uses
Single-agent treatment of both untreated patients and alpha-interferon–refractory hairy cell leukemia (HCL) patients with active disease as defined by clinically significant anemia, neutropenia, thrombocytopenia, or disease-related symptoms.
Unlabeled uses:
Treatment of chronic lymphocytic leukemia, prolymphocytic leukemia, non-Hodgkin’s lymphoma, cutaneous T-cell lymphoma, and peripheral T-cell lymphomas.
Contraindications
Hypersensitivity to pentostatin; see Drug/Lab Interactions.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Assess drug profile before administration. ■ Severe renal, liver, pulmonary, and CNS toxicities have occurred at higher doses; do not exceed recommended dose. ■ Usually administered by or under the supervision of a physician specialist. ■ Myelosuppression, especially neutropenia, is most severe during the first few courses of treatment. ■ Must consider risk/benefit in patients with some bone marrow suppression, the possibility of chickenpox or herpes zoster, a history of gout or urate renal stones, renal function impairment, or previous cytotoxic drug or radiation therapy. Use extreme caution. ■ After 6 months of treatment, assess for response; if partial or complete response is not evident, discontinue treatment. If partial response is evident, re-evaluate as indicated but do not treat beyond 12 months.
Monitor:
Monitor CBC (including differential and platelet count) and SCr before each dose and as indicated. Blood chemistries, including serum uric acid, and a CrCl assay are required before and during treatment. ■ Prehydration with 500 to 1,000 mL D5/1/2NS or an equivalent is required. An additional 500 mL is required postadministration. ■ Treatment of patients with infection may exacerbate symptoms and cause death. Control infection before treatment is initiated. Withhold treatment if an active infection occurs; resume when infection is controlled. ■ Prophylactic antiemetics recommended (e.g., prochlorperazine [Compazine], ondansetron [Zofran]); continue for 48 to 72 hours. ■ Observe closely for severe rashes, nervous system toxicity, and myelosuppression (especially after initial cycles); pentostatin may have to be withheld or discontinued. ■ For severe neutropenia beyond the initial cycles, evaluate for disease status, including a bone marrow examination. ■ Assess response to treatment with periodic monitoring of peripheral blood for hairy cells. Bone marrow aspirates and biopsies may be required at 2- to 3-month intervals. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood).
Patient education:
Consider birth control options; nonhormonal birth control recommended. ■ Report rashes, symptoms of infection, or bruising and bleeding immediately. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy; can cause fetal harm. ■ Discontinue breast-feeding. ■ Safety for use in pediatric patients under 18 years of age not established.
Elderly:
Consider decreased renal function.
Drug/lab interactions
Assess drug profile before administration. ■ Do not use with fludarabine (Fludara); may increase risk of fatal pulmonary toxicity. ■ Combination therapy with carmustine (BiCNU), etoposide (VePesid), and high-dose cyclophosphamide as part of the ablative regimen for bone marrow transplant has caused acute pulmonary edema and hypotension. Deaths have occurred. ■ Pentostatin enhances the effects of vidarabine (Ara-A). Combined use of these agents may result in an increase in adverse reactions associated with each drug. The therapeutic benefit of this drug combination has not been established. ■ May cause skin rash with allopurinol. ■ Elevates liver function tests; usually reversible. ■ Do not administer live virus vaccines to patients receiving antineoplastic drugs. ■ Uric acid levels may increase; increased dose of gout agents (e.g., colchicine, probenecid, sulfinpyrazone [Anturane]) may be indicated. ■ Leukopenia and thrombocytopenia increased by agents causing blood dyscrasias (e.g., anticonvulsants [phenytoin (Dilantin)], penicillins, phenothiazines, and many others).
Side effects
Anemia, anorexia, chills, cough, diarrhea, fatigue, fever, GU disorders, headache, hepatic disorders/elevated liver function tests, hypersensitivity reactions, infection, leukopenia, lung disorders, myalgia, nausea, neurologic disorders/CNS, pain, rashes, skin disorders, thrombocytopenia, upper respiratory infections, and vomiting occur in 10% of patients and may require discontinuation of treatment. Abdominal pain; abnormal ECG; abnormal thinking; abnormal vision; anxiety; arthralgia; asthenia; back pain; bronchitis; cardiac arrhythmias; chest pain; confusion; conjunctivitis; constipation; depression; dizziness; dry skin; dyspnea; dysuria; ear pain; ecchymosis; eczema; elevated BUN, creatinine, and LDH; epistaxis; eye pain; flatulence; flu syndrome; hematuria; hemorrhage; herpes simplex; herpes zoster; insomnia; lung edema; lymphadenopathy; maculopapular rash; malaise; neoplasm; nervousness; paresthesia; peripheral edema; petechia; pharyngitis; pneumonia; pruritus; rhinitis; seborrhea; sinusitis; skin discoloration; somnolence; stomatitis; sweating; thrombophlebitis; vesiculobullous rash; weight loss; and death have occurred in 3% to 10% or more of patients.
Antidote
Keep physician informed of all side effects; most will be treated symptomatically if indicated. Withhold dose and notify physician for elevated SCr, absolute neutrophil count below 200 cells/mm3, myelosuppression, infection, CNS toxicity, or severe rash. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. Overdose may cause death due to severe renal, hepatic, pulmonary, or CNS toxicity. There is no specific antidote. Supportive therapy as indicated will help sustain the patient.
Peramivir
(per-AM-i-vir)
Rapivab
Antiviral
(Influenza virus neuraminidase inhibitor)
pH 5.5 to 8.5
Usual dose
Administer peramivir within 2 days of onset of symptoms of influenza.
A single dose of 600 mg administered as an IV infusion over 15 to 30 minutes.
Dose adjustments
No dose adjustment is required in patients with a creatinine clearance of 50 mL/min or higher. ■ Reduce dose in patients with a baseline creatinine clearance below 50 mL/min using the recommendations in the following chart.
| Dose Adjustment of Peramivir in Impaired Renal Function | |
| Creatinine Clearance (mL/min) | Recommended Dose (mg) |
| ≥50 mL/min | 600 mg |
| 30 to 49 mL/min | 200 mg |
| 10 to 29 mL/min | 100 mg |
| Patients with chronic renal impairment maintained on hemodialysis | Administer after dialysis at a dose adjusted based on renal function. |
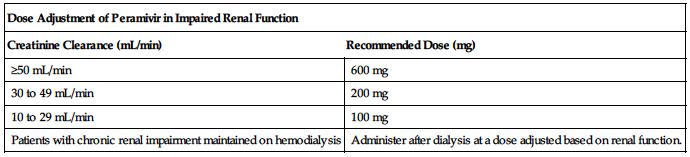
■ No dose adjustment is required based on gender, impaired hepatic function, weight, or race (was evaluated primarily in Asians and Caucasians).
Dilution
Available in single-use vials containing 200 mg/ 20 mL (10 mg/mL) as a clear, colorless solution. Contains no preservatives. Do not use if seal over bottle opening is broken or missing. Calculate the number of vial(s) required for the recommended dose, and withdraw the required volume of peramivir and dilute by transferring into an IV bag or container of NS, 1/2NS, D5W, or LR to a maximum volume of 100 mL.
Filters:
No data available from manufacturer.
Storage:
Before use, store in original cartons at CRT. Administer fully diluted solutions immediately or refrigerate at 2° to 8° C (36° to 46° F) for up to 24 hours. If refrigerated, allow the diluted solution to reach room temperature, then administer immediately. Discard any unused diluted solution of peramivir after 24 hours.
Compatibility
Compatible with NS, 1/2NS, D5W, or LR. Also compatible with materials commonly used for administration such as polyvinylchloride (PVC) bags and PVC-free bags, polypropylene syringes, and polyethylene tubing. Manufacturer states, “Do not mix or co-infuse with other intravenous medications.”
Rate of administration
A single dose as an infusion equally distributed over 15 to 30 minutes.
Actions
Peramivir is an antiviral drug with activity against the influenza virus. It is an inhibitor of the influenza virus neuraminidase, an enzyme that releases viral particles from the plasma membrane of infected cells. The relationship between the antiviral activity in cell culture, the inhibitory activity in the neuraminidase assay, and the inhibition of influenza virus replication in humans has not been established. Maximum serum concentration is reached by the end of the infusion. In vitro binding to human plasma proteins is less than 30%. Not significantly metabolized in humans. Elimination half-life is approximately 20 hours. Primarily excreted as unchanged drug in urine.
Indications and uses
Treatment of acute uncomplicated influenza in patients 18 years or age and older who have been symptomatic for no more than 2 days.
Limitations of use:
Efficacy of peramivir is based on clinical trials of naturally occurring influenza in which the predominant influenza infections were influenza A virus; a limited number of subjects infected with influenza B virus were enrolled. ■ Influenza viruses change over time. Emergence of resistance substitutions could decrease drug effectiveness. Other factors (e.g., changes in viral virulence) might also diminish the clinical benefit of antiviral drugs. Prescribers should consider available information on influenza drug susceptibility patterns and treatment effects when deciding whether to use peramivir. ■ The use of peramivir was not shown to provide benefit to patients with serious influenza requiring hospitalization.
Contraindications
Manufacturer states, “None.”
Precautions
Rare cases of serious skin and hypersensitivity reactions, including erythema multiforme, have been reported. ■ Influenza can be associated with a variety of neurologic and behavioral symptoms that can include events such as abnormal behavior, delirium, and hallucinations, which in some cases result in fatal outcomes. These events may occur in the setting of encephalitis or encephalopathy but also can occur in uncomplicated influenza. Neuropsychiatric events (e.g., delirium and abnormal behavior) have been reported in patients with influenza who are receiving neuraminidase inhibitors, including peramivir. The contribution of peramivir to these events has not been established. ■ There is no evidence for the efficacy of peramivir in any illness caused by agents other than influenza viruses. Serious bacterial infections may begin with influenza-like symptoms or may coexist with or occur as complications during the course of influenza. Peramivir has not been shown to prevent such complications. ■ Circulating seasonal influenza strains expressing neuraminidase resistance–associated substitutions have been observed in individuals who have not received peramivir. Prescribers should consider available information from the CDC on influenza virus drug susceptibility patterns and treatment effects when deciding whether to use peramivir. ■ Has not been studied in patients with impaired hepatic function; however, because peramivir is cleared in urine by glomerular filtration, no clinically relevant problems are expected. ■ Cross-resistance between peramivir (Rapivab), oseltamivir (Tamiflu), and zanamivir (Relenza) was observed in neuraminidase biochemical assays and cell culture assays. The clinical impact of this reduced susceptibility is unknown.
Monitor:
Monitor for serious skin reactions. Appropriate treatment should be instituted if a serious skin reaction occurs or is suspected. ■ Monitor for potential secondary bacterial infections and treat with antibiotics as appropriate. ■ Monitor for S/S of a hypersensitivity reaction (e.g., hypotension, rash, urticaria, tightness of the chest, wheezing). ■ Patients with influenza should be closely monitored for signs of abnormal behavior.
Patient education:
There is a risk of serious skin reactions. Seek immediate medical attention if a skin reaction occurs. ■ There is a risk of neuropsychiatric events in patients with influenza. Patients should contact their physician if they experience signs of abnormal behavior after receiving peramivir. ■ Promptly report S/S of a hypersensitivity reaction (e.g., hives, rash, shortness of breath, wheezing).
Maternal/child:
Category C: use during pregnancy only if clearly needed. ■ It is not known whether peramivir is excreted in human milk; use caution during breast-feeding. ■ Safety and effectiveness in pediatric patients under 18 years of age have not been established. ■ In a single-arm trial conducted in Japan, pediatric patients with uncomplicated influenza (28 days to 16 years of age) were treated with a single dose of peramivir 10 mg/kg. The most common clinical and laboratory adverse events included decreased neutrophil count, diarrhea, and vomiting.
Elderly:
Numbers in clinical studies were insufficient to determine if elderly patients respond differently than do younger subjects.
Drug/lab interactions
The concurrent use of peramivir with live attenuated influenza vaccine (LAIV) intranasal has not been evaluated. Because of the potential for interference between these two products, avoid the use of LAIV within 2 weeks before or 48 hours after administration of peramivir unless medically indicated. For LAIV, antiviral drugs may inhibit viral replication and may reduce vaccine efficacy. ■ Inactivated influenza vaccine can be administered at any time. ■ Not a substrate for CYP enzymes, does not affect glucuronidation, and is not a substrate or inhibitor of P-glycoprotein–mediated transport. ■ No evidence of drug-drug interactions when peramivir was administered with oral rimantadine (Flumadine), oral oseltamivir (Tamiflu), or oral contraceptives containing ethinyl estradiol and levonorgestrel or when peramivir IM was administered with oral probenecid.
Side effects
The most common adverse reaction is diarrhea. Serious skin and hypersensitivity reactions and neuropsychiatric events have occurred. Constipation; decreased neutrophils; hypertension; increased ALT, AST, creatine phosphokinase, and serum glucose; and insomnia were also reported.
Post-marketing:
Abnormal behavior, exfoliative dermatitis, hallucination, rash, and Stevens-Johnson syndrome.
Antidote
Notify physician of any side effects. Discontinue the drug if indicated. Treat overdose with general supportive measures, including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote. Treat hypersensitivity reactions as indicated (e.g., diphenhydramine [Benadryl], epinephrine [Adrenalin], albuterol) and resuscitate as necessary. Removed by hemodialysis.
Pertuzumab 
(per-TOOZ-ue-mab)
Perjeta
Recombinant monoclonal antibody
Antineoplastic
pH 6
Usual dose
Preassessment is required; see Monitor. Pertuzumab is given in combination with trastuzumab (Herceptin) and docetaxel (Taxotere). See docetaxel and trastuzumab monographs; preassessment and premedication indicated. Pertuzumab, trastuzumab, and docetaxel should be administered sequentially. Pertuzumab and trastuzumab can be given in any order. Docetaxel should be administered after pertuzumab and trastuzumab infusions are complete. An observation period of 30 to 60 minutes is recommended after each pertuzumab infusion and before beginning any subsequent infusion of trastuzumab or docetaxel.
Metastatic breast cancer (MBC)
Initial doses:
Pertuzumab:
840 mg as an infusion over 60 minutes; see Monitor; preassessment required.
Trastuzumub:
8 mg/kg as an infusion over 90 minutes.
Docetaxel:
75 mg/M2 as an infusion over 60 minutes.
Subsequent doses:
Begin 3 weeks after initial doses.
Pertuzumab:
420 mg administered as an infusion over 30 to 60 minutes and repeated every 3 weeks.
Trastuzumub:
6 mg/kg as an infusion over 30 to 90 minutes and repeated every 3 weeks.
Docetaxel:
If the initial dose is well tolerated, the dose may be escalated to 100 mg/M2 and repeated every 3 weeks.
Neoadjuvant treatment of breast cancer
Administer pertuzumab every 3 weeks for 3 to 6 cycles as part of one of the following treatment regimens for early breast cancer. Actual initial and subsequent doses of pertuzumab, trastuzumab, and docetaxel are the same as above in MBC. Note all comments under Usual Dose.
• Four preoperative cycles of pertuzumab in combination with trastuzumab and docetaxel followed by 3 postoperative cycles of fluorouracil, epirubicin, and cyclophosphamide (FEC).
• Three preoperative cycles of FEC alone followed by 3 preoperative cycles of pertuzumab in combination with docetaxel and trastuzumab.
• Six preoperative cycles of pertuzumab in combination with docetaxel, carboplatin, and trastuzumab (TCH) (escalation of docetaxel above 75 mg/M2 is not recommended).
Following surgery, patients should continue to receive trastuzumab to complete 1 year of treatment.
Dose adjustments
If a dose is delayed or missed and the time between two infusions is less than 6 weeks, administer the 420-mg dose. Do not wait until the next planned dose. ■ If the time between two infusions is 6 weeks or more, readminister the initial dose of 840 mg and follow with the subsequent dose regimen of 420 mg beginning in 3 weeks. ■ Discontinue immediately for a serious hypersensitivity reaction. ■ Withhold pertuzumab and trastuzumab for at least 3 weeks if there is a drop in left ventricular ejection fraction (LVEF) to less than 45% or a LVEF of 45% to 49% with a 10% or greater absolute decrease below pretreatment values, and reassess LVEF in 3 weeks. Pertuzumab may be resumed if the LVEF recovers to greater than 49%, or if it recovers to between 45% and 49% if associated with less than a 10% absolute decrease below pretreatment values. If after a repeat assessment within approximately 3 weeks the LVEF has not improved or has declined further, pertuzumab and trastuzumab should be discontinued unless benefits outweigh risks for the individual patient. ■ Pertuzumab should be discontinued if trastuzumab treatment is discontinued. ■ Dose reductions are not recommended for pertuzumab. ■ See docetaxel monograph for docetaxel dose adjustments. ■ No dose adjustment is indicated in patients with mild or moderate renal impairment. In patients with severe renal impairment (CrCl less than 30 mL/min), no dose adjustments can be recommended because of the limited pharmacokinetic data available. ■ See Rate of Administration, Precautions, Monitor, and Antidote.
Dilution
Withdraw the calculated dose of pertuzumab from the vial(s) and inject into a 250-mL PVC or non-PVC polyolefin infusion bag of NS. Invert diluted solution gently to mix. Do not shake. Dilute only with NS. Do not use D5W. Immediate use is preferred.
Filters:
Not required by manufacturer; additional data not available.
Storage:
Store vials in carton in refrigerator at 2° to 8° C (36° to 46° F). Do not freeze. Do not shake. Protect from light. Immediate use preferred; if the diluted solution is not used immediately, however, it can be refrigerated for 24 hours.
Compatibility
Manufacturer states, “Do not mix pertuzumab with other drugs” and lists as incompatible with D5W.
Rate of administration
For IV infusion only; do not administer as an IV push or IV bolus. The rate of infusion may be slowed or interrupted if the patient develops an infusion-associated reaction. Discontinue immediately for a serious hypersensitivity reaction.
Pertuzumab:
Administer the initial dose over 60 minutes. Administer subsequent doses over 30 to 60 minutes. An observation period of 30 to 60 minutes is recommended after each pertuzumab infusion and before beginning any subsequent infusion of trastuzumab or docetaxel.
Trastuzumab:
Administer the initial dose over 90 minutes. Administer subsequent doses over 30 to 90 minutes. May be administered before or after pertuzumab.
Docetaxel:
Each dose equally distributed over 60 minutes. Administer after pertuzumab and trastuzumab infusions have been completed.
Actions
A recombinant humanized monoclonal antibody. It targets the human epidermal growth factor receptor 2 protein (HER2) and blocks ligand-dependent heterodimerization of HER2 with other HER family members, including EGFR, HER3, and HER4. By inhibiting specific pathways, cell growth arrest and apoptosis can occur. Pertuzumab also mediates antibody-dependent, cell-mediated cytotoxicity. Using pertuzumab alone inhibits the proliferation of human tumor cells, whereas the combination of pertuzumab and trastuzumab greatly augments antitumor activity in HER2-overexpressing xenograft models. Steady-state concentration can be reached after the first maintenance dose.
Indications and uses
Pertuzumab in combination with trastuzumab and docetaxel is indicated for the treatment of patients with HER2-positive metastatic breast cancer who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease. ■ Pertuzumab in combination with trastuzumab and docetaxel is indicated for the neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or early-stage breast cancer (either greater than 2 cm in diameter or node positive) as part of a complete treatment regimen for early breast cancer. This indication is based on demonstration of an improvement in pathologic complete response rate. Data demonstrating improvement in event-free survival or overall survival is not available.
Limitations of use:
The safety of pertuzumab as part of a doxorubicin-containing regimen has not been established. ■ The safety of pertuzumab administered for more than 6 cycles for early breast cancer has not been established.
Contraindications
Known hypersensitivity to pertuzumab or to any of its excipients.
Precautions
For IV infusion only; do not administer as an IV push or IV bolus. ■ Assess HER2 status before beginning pertuzumab therapy. Testing should be done by a laboratory with demonstrated proficiency in the testing process using FDA-approved tests. In clinical studies, the only patients who have received benefit from pertuzumab therapy are those with HER2 protein overexpression. ■ Can cause fetal harm when administered to pregnant women; see Patient Education and Maternal/Child. ■ Administered by or under the direction of a physician specialist. ■ Adequate laboratory and supportive medical resources must be available. ■ Emergency equipment and drugs for treatment of left ventricular dysfunction and/or hypersensitivity or infusion reactions must be immediately available; see Antidote. ■ Can result in subclinical and clinical cardiac failure. Evaluate left ventricular function in all patients before and during treatment with pertuzumab. Decreases in LVEF with drugs that block HER2 activity, including pertuzumab, have been reported. Patients who have received prior anthracyclines or radiotherapy to the chest wall may be at higher risk for decreased LVEF. ■ Hypersensitivity reactions, including anaphylaxis, and infusion reactions have occurred. ■ Trastuzumab and docetaxel also have extensive precautions and monitoring requirements; a review of their monographs is imperative. ■ Effects on patients with severe renal impairment and/or hepatic impairment have not been studied. ■ A protein substance; has the potential for immunogenicity.
Monitor:
Verify pregnancy status before beginning pertuzumab therapy ■ Assess LVEF before beginning therapy and at regular intervals during treatment (e.g., every 3 months in the metastatic setting and every 6 weeks in the neoadjuvant setting) to verify that it remains within expected parameters; see Dose Adjustments. ■ Monitor for S/S of a hypersensitivity reaction (e.g., chest pain, chills, dizziness, dyspnea, fever, flushing, hypotension, nausea, pruritus, rash, urticaria) or infusion reaction (e.g., asthenia, chills, dysgeusia, fatigue, fever, headache, myalgia, vomiting). Observe closely for 60 minutes after the first infusion and for at least 30 minutes after subsequent infusions. ■ Trastuzumab and docetaxel have additional monitoring requirements.
Patient education:
Avoid pregnancy; effective contraception required while receiving therapy and for 7 months following the last dose. ■ During infusion, promptly report chills and/or fever and other S/S of an infusion or hypersensitivity reaction (e.g., chest pain, dizziness, feeling faint, flushing, hives, itching, nausea, pruritus, rash, shortness of breath). ■ Promptly report new onset or worsening of shortness of breath, cough, swelling of ankles/legs, swelling of the face, palpitations, weight gain of more than 5 pounds in 24 hours, dizziness, or loss of consciousness. ■ Report a suspected pregnancy immediately. If a pregnancy occurs during therapy, if pertuzumab is administered during a pregnancy, or if pregnancy occurs within 7 months of the last dose of pertuzumab, report exposure immediately to the Genentech Adverse Event Line. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy; may cause fetal harm. Exposure can result in embryo-fetal death and birth defects. Studies in animals have resulted in oligohydramnios, delayed renal development, and embryo-fetal deaths; see Patient Education. ■ Encourage patients who are exposed during pregnancy or who become pregnant within 7 months after the last dose of pertuzumab to enroll in the MotHER Pregnancy Registry. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
No differences in safety and effectiveness noted compared with younger adults.
Drug/lab interactions
No formal drug interaction studies have been done. ■ No drug-to-drug interactions observed between pertuzumab and trastuzumab or between pertuzumab and docetaxel.
Side effects
The most common side effects when pertuzumab was administered in combination with trastuzumab and docetaxel were alopecia, diarrhea, fatigue, nausea, neutropenia, peripheral neuropathy, and rash. Anemia, thrombocytopenia, and vomiting were reported when carboplatin was added to the regimen. The most common Grade 3 and 4 adverse reactions were anemia, asthenia, diarrhea, fatigue, febrile neutropenia, leukopenia, neutropenia, and peripheral neuropathy. Adverse events occurring in 10% or more of patients treated with pertuzumab include arthralgia, constipation, decreased appetite, dizziness, dry skin, dysgeusia, dyspnea, headache, increased lacrimation, insomnia, itching, mucosal inflammation, myalgia, nail disorders, nasopharyngitis, stomatitis, upper respiratory tract infections, and vomiting. Clinically relevant adverse events reported in fewer than 10% of patients include left ventricular dysfunction (including symptomatic left ventricular systolic dysfunction), hypersensitivity and/or infusion reactions, paronychia, and pleural effusion. Adverse reactions were reported less frequently after discontinuation of docetaxel treatment.
Antidote
Notify physician of all side effects. Most will be treated symptomatically. If signs of an infusion reaction occur, slow or interrupt the infusion and treat appropriately. Monitor patients carefully until symptoms resolve. Discontinue immediately for a serious hypersensitivity reaction. Discontinue pertuzumab for a confirmed clinically significant decrease in left ventricular function. If treatment with pertuzumab and trastuzumab has been withheld because of a drop in LVEF and if LVEF has not improved or has declined further after a repeat assessment within approximately 3 weeks, pertuzumab and trastuzumab should be discontinued unless benefits outweigh risks for the individual patient. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity from concurrent antineoplastics. Treat hypersensitivity reactions with epinephrine, antihistamines, corticosteroids, bronchodilators, and oxygen. Resuscitate as indicated.
Phenobarbital sodium
(fee-no-BAR-bih-tal SO-dee-um)
Luminal Sodium
Barbiturate
Sedative-hypnotic
Anticonvulsant
pH 8.5 to 10.5
Usual dose
Use only enough medication to achieve the desired effect. May take up to 15 minutes to reach peak levels in the brain; guard against overdose and excessive respiratory depression.
Hypnotic:
100 to 325 mg.
Sedative:
30 to 120 mg/day in 2 or 3 divided doses (15 to 60 mg every 12 hours or 10 to 40 mg every 8 hours).
Anticonvulsant:
200 to 320 mg. May be repeated if necessary. Maximum dose usually does not exceed 600 mg.
Status epilepticus:
Loading dose:
10 to 20 mg/kg in single or divided doses. May give an additional 5 mg/kg every 15 to 30 minutes up to a maximum dose of 30 mg/kg.
Maintenance dose:
1 to 3 mg/kg/24 hr or 0.5 to 1.5 mg/kg every 12 hours.
Pediatric dose
See comments under Usual Dose.
Preoperative sedation:
1 to 3 mg/kg of body weight 60 to 90 minutes before procedure.
Status epilepticus:
Loading dose:
15 to 18 mg/kg as a single dose or in divided doses. May give an additional 5 mg/kg every 15 to 30 minutes up to a maximum total dose of 30 mg/kg.
Maintenance dose: Infants:
2.5 to 3 mg/kg every 12 hours.
Ages 1 to 5:
3 to 4 mg/kg every 12 hours.
Ages 6 to 12:
2 to 3 mg/kg every 12 hours.
Over 12 years of age:
0.5 to 1.5 mg/kg every 12 hours. Up to 12 mg/kg/24 hours has been used in maintenance doses.
Neonatal dose
See comments under Usual Dose.
Status epilepticus:
Loading dose:
15 to 20 mg/kg as a single dose or in divided doses.
Maintenance dose:
1.5 to 2 mg/kg every 12 hours; may be increased to 2.5 mg/kg every 12 hours if needed. Therapeutic range is 15 to 40 mg/L. Because of its long half-life, it may take 2 to 3 weeks to reach steady-state levels.
Dose adjustments
Reduce dose in impaired renal or hepatic function; usually required in the debilitated or elderly. ■ See Drug/Lab Interactions.
Dilution
Sterile powder must be slowly diluted with SWFI. Use a minimum of 3 mL of diluent. Also available in sterile vials and tubexes. Best if further diluted up to 10 mL with SWFI. Solutions from powder form must be freshly prepared. Use only absolutely clear solutions. Discard powder or solution exposed to air for 30 minutes.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Amikacin, aminophylline, calcium chloride, calcium gluconate, colistimethate (Coly-Mycin M), dimenhydrinate, meropenem (Merrem IV), verapamil.
Y-site:
Doripenem (Doribax), doxapram (Dopram), enalaprilat (Vasotec IV), fentanyl, fosphenytoin (Cerebyx), hydromorphone (Dilaudid), levofloxacin (Levaquin), linezolid (Zyvox), meropenem (Merrem IV), methadone (Dolophine), morphine, propofol (Diprivan).
Rate of administration
60 mg (gr 1) or fraction thereof over 1 minute. Titrate slowly to desired effect. Rapid injection rate may cause symptoms of overdose (e.g., serious respiratory depression).
Status epilepticus:
A single loading dose over 10 to 15 minutes.
Actions
A sedative, hypnotic barbiturate of long duration with potent anticonvulsant effects. Phenobarbital is a CNS depressant. Onset of action is prompt by the IV route and becomes rapidly more intense. Effects last from 6 to 10 hours. Will effectively depress the motor cortex with small doses. Pain perception is unimpaired. Rapidly absorbed by all body tissues and excreted in changed form in the urine. Excreted more readily in alkaline urine. Crosses the placental barrier. Secreted in breast milk.
Indications and uses
Prolonged sedation (medical and psychiatric). ■ Anticonvulsant.
Contraindications
History of porphyria, impaired renal function, impaired hepatic function especially with any signs of hepatic coma, known hypersensitivity to barbiturates, previous addiction, severe respiratory depression including dyspnea, obstruction, or cor pulmonale.
Precautions
IV route usually reserved for critical situations. ■ Use caution in elderly and debilitated patients and those with asthma, pulmonary disease, shock, and impaired renal or hepatic function. ■ Status epilepticus can occur from too-rapid withdrawal. ■ May be habit forming. Use caution in acute or chronic pain. ■ Benzodiazepines (diazepam [Valium], midazolam [Versed]) generally preferred for sedation.
Monitor:
Keep patient under constant observation. Record vital signs every hour, or more often if indicated. ■ Maintain a patent airway. ■ Monitor hematopoietic, renal and hepatic systems in any extended therapy. ■ Treat the cause of a convulsion. ■ Keep equipment for artificial ventilation available. ■ Highly alkaline. Determine absolute patency of vein; use of large veins preferred to prevent thrombosis. Avoid extravasation. Intra-arterial injection will cause gangrene. ■ Monitor serum levels as indicated; the therapeutic range in adults is 20 to 40 mcg/mL (15 to 40 mcg/mL in pediatric patients). Because of its long half-life, it may take 2 to 3 weeks to reach steady-state levels. ■ See Drug/Lab Interactions.
Patient education:
Avoid alcohol or other CNS depressants (e.g., antihistamines, diazepam [Valium]). May be habit forming. ■ May require alternate birth control.
Maternal/child:
Category D: avoid pregnancy; will cause birth defects. ■ May cause drowsiness in the nursing infant. ■ See Precautions.
Elderly:
See Dose Adjustments and Precautions. ■ Often have increased sensitivity to barbiturates; may cause marked excitement, depression, confusion, and increased risk of barbiturate-induced hypothermia. ■ Consider age-related hepatic or renal impairment and concomitant disease or drug therapy.
Drug/lab interactions
Use extreme caution if any other CNS depressants have been given, such as alcohol, aminoglycoside antibiotics, narcotic analgesics, anesthetics, antidepressants, antihistamines, hypnotics, MAO inhibitors, phenothiazines, sedatives, tranquilizers. Potentiation with respiratory depression may occur. ■ Inhibits effectiveness of corticosteroids, doxycycline, oral anticoagulants, oral contraceptives, propranolol, quinidine, and theophylline. Capable of innumerable interactions with many drugs. ■ May increase orthostatic hypotension with furosemide (Lasix). ■ Monitor phenytoin (Dilantin), felbamate (Felbatol), carbamazepine (Tegretol), valproic acid and phenobarbital levels when any combination of these drugs is used concurrently. ■ May decrease the pharmacologic effect of vitamin D. ■ May decrease plasma concentrations and effectiveness of triazole antifungals (e.g., itraconazole [Sporanox]).
Side effects
Rarely occur with slow injection of average doses.
Average dose:
Depression, dermatitis, facial edema, fever, headache, hypotension, nausea, neonatal apnea, respiratory depression (hypoventilation), thrombocytopenic purpura, vertigo.
Overdose:
Apnea, coma, cough reflex depression, delirium, flat EEG (reversible unless hypoxic damage has occurred), hypotension, laryngospasm, lowered body temperature, pulmonary edema, renal shutdown, respiratory depression, sluggish or absent reflexes, stupor.
Antidote
Notify the physician of any side effects. Symptomatic and supportive treatment is most important in overdose. Maintain an adequate airway with artificial ventilation if indicated. Keep the patient warm. IV volume expanders (dextran) and other IV fluids will help maintain adequate circulation. Diuretics may promote the elimination of the drug. Vasopressors (e.g., dopamine) will maintain BP.
Phenylephrine hydrochloride 
(fen-ill-EF-rin hy-droh-KLOR-eyed)
Neo-Synephrine, Vazculep
Vasopressor
pH 3 to 6.5
Usual dose
Perioperative setting:
Bolus:
40 to 100 mcg by IV bolus administration. May repeat every 1 to 2 minutes as needed, not to exceed a total dose of 200 mcg.
A second manufacturer lists an initial bolus of 50 to 100 mcg with a range of 50 to 250 mcg.
Continuous infusion:
If BP is below target goal, begin a continuous infusion of 10 to 35 mcg/min, not to exceed 200 mcg/min.
A second manufacturer lists a rate of 0.5 to 1.4 mcg/kg/min. Titrate to blood pressure goal.
Septic or other vasodilatory shock (no longer recommended for routine use for this indication):
Do not administer a bolus. Begin with a continuous infusion of 0.5 to 6 mcg/kg/min. Titrate to blood pressure goal. Doses above 6 mcg/kg/min do not show a significant incremental increase in blood pressure.
Another source recommends:
Begin infusion at 100 to 180 mcg/min (0.1 to 0.18 mg/min) until BP is stabilized at a low normal for specific individual. Maintain with 40 to 60 mcg/min (0.04 to 0.06 mg/min). Titrate to desired effect.
Dose adjustments
Patients with liver cirrhosis (Child-Pugh Class B and Class C) may have decreased responsiveness to phenylephrine. Higher-end doses may be required to achieve BP goal. ■ Patients with end-stage renal disease may have increased responsiveness to phenylephrine. Initiate dosing at lower end of dosing range.
Dilution
IV bolus:
Dilute 10 mg (1 mL of a 10 mg/mL solution) with 99 mL of NS or D5W to provide a final concentration of 100 mcg/mL. Withdraw an appropriate dose from this solution prior to bolus administration.
Infusion:
Dilute 10 mg in 500 mL of NS or D5W to provide a final concentration of 20 mcg/mL.
Storage:
Store unopened vials at CRT in carton. Protect from light. Diluted solution should not be held for more than 4 hours at RT or for more than 24 hours under refrigeration. Discard any unused portion.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Chloramphenicol (Chloromycetin), dobutamine, lidocaine, potassium chloride (KCl), sodium bicarbonate.
Y-site:
Amiodarone (Nexterone), anidulafungin (Eraxis), argatroban, bivalirudin (Angiomax), caspofungin (Cancidas), cisatracurium (Nimbex), dexmedetomidine (Precedex), doripenem (Doribax), etomidate (Amidate), famotidine (Pepcid IV), fenoldopam (Corlopam), hetastarch in electrolytes (Hextend), levofloxacin (Levaquin), micafungin (Mycamine), propofol (Diprivan), remifentanil (Ultiva), telavancin (Vibativ), vasopressin, zidovudine (AZT, Retrovir).
Rate of administration
IV injection:
Infusion:
See Usual Dose. Titrate to maintain individual’s low-normal BP. Use an infusion pump or microdrip (60 gtt/mL) to administer. Central line preferred.
Actions
An alpha-1 adrenergic receptor agonist. Interacts with the receptors on the vascular smooth muscle cells, resulting in vasoconstriction. Increases in systolic blood pressure, diastolic blood pressure, mean arterial blood pressure, and total peripheral vascular resistance are observed within minutes of administration. As blood pressure increases, vagal activity also increases, resulting in a reflex bradycardia. Active on most vascular beds, including renal, pulmonary, and splanchnic arteries. Duration of effect is 15 to 20 minutes. Terminal half-life is 2.5 hours. Metabolized primarily by monoamine oxidase and sulfotransferase. Excreted in urine, primarily as inactive metabolites.
Indications and uses
Treatment of clinically important hypotension resulting primarily from vasodilation in settings such as septic shock or anesthesia. (No longer recommended for routine use in treatment of hypotension related to septic shock.)
Contraindications
Hypersensitivity to phenylephrine or any of its components.
Precautions
Usually administered by or under the supervision of a physician knowledgeable in its use. ■ Intravascular volume depletion should be corrected. ■ Correct acidosis. Acidosis may reduce effectiveness of phenylephrine. ■ Because of its pressor effects, phenylephrine can precipitate angina in patients with severe arteriosclerosis or history of angina, exacerbate underlying heart failure, and increase pulmonary arterial pressure. ■ Avoid extravasation. Can cause necrosis or sloughing of tissue. ■ Can cause peripheral and visceral vasoconstriction and ischemia to vital organs, particularly in patients with extensive peripheral vascular disease. ■ Can cause severe bradycardia and decreased cardiac output. ■ Can increase the need for renal replacement therapy in patients with septic shock. ■ The pressor response to adrenergic drugs, including phenylephrine, can be increased in patients with autonomic dysfunction, such as may occur with spinal cord injuries. ■ Contains bisulfites; use caution in allergic individuals. ■ See Drug/Lab Interactions.
Monitor:
Monitor vital signs. ■ Check infusion site for free flow. Discontinue IV administration if vein infiltrates or is thrombosed. ■ Monitor renal function. ■ See Precautions, Drug/Lab Interactions.
Maternal/child:
Category C: safety for use in pregnancy or breast-feeding not established. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
See Precautions; may have increased sensitivity to effects.
Drug/lab interactions
Oxytocic drugs (e.g., oxytocin) potentiate the increasing blood pressure effects of sympathomimetic pressor amines, including phenylephrine, with the potential for hemorrhagic stroke. ■ The pressor effect of phenylephrine is also increased in patients receiving MAO inhibitors (e.g., selegiline [Eldepryl]), tricyclic antidepressants (e.g., desipramine [Norpramin]), angiotensin, aldosterone, atropine, steroids (e.g., hydrocortisone), norepinephrine transporter inhibitors (e.g., atomoxetine), ergot alkaloids (e.g., methylergonovine maleate [Methergine]). ■ The pressor effect of phenylephrine is decreased in patients receiving alpha-adrenergic antagonists (e.g., doxazosin [Cardura], prazosin [Minipress]), phosphodiesterase type 5 inhibitors (e.g., sildenafil [Viagra]), mixed alpha- and beta-receptor antagonists (e.g., labetalol [Trandate], carvedilol [Coreg]), calcium channel blockers (e.g., diltiazem [Cardizem], nifedipine [Procardia]), benzodiazepines (e.g., diazepam [Valium], midazolam [Versed]), ACE inhibitors (e.g., enalapril [Vasotec], lisinopril [Zestril, Prinivil]), centrally acting sympatholytic agents (e.g., reserpine, guanfacine [Intuniv]).
Side effects
The most common side effects are headache, nausea, and vomiting. Other reported side effects include arrhythmias, blurred vision, chest pain, diaphoresis, dyspnea, epigastric pain, extravasation, fullness of head, hypersensitivity (sulfite sensitivity), hypertension, hypertensive crisis, ischemia, lower cardiac output, neck pain, nervousness, paresthesia, pruritus, pulmonary edema, rales, reflexive bradycardia, skin blanching, skin necrosis with extravasation, and tremor.
Antidote
To prevent sloughing and necrosis in areas of extravasation, with a fine hypodermic needle inject 5 to 10 mg of phentolamine (Regitine) diluted in 10 to 15 mL of NS liberally throughout the tissue in the extravasated area. Treatment should be started as soon as extravasation is recognized. Notify the physician of all side effects. IM injection may be preferable. Treat hypertension with phentolamine (Regitine). Treat cardiac arrhythmias as indicated. Treat bradycardia with atropine. Resuscitate as necessary.
Phenytoin sodium 
(FEN-ih-toyn SO-dee-um)
Dilantin
Hydantoin
Anticonvulsant
pH 12
Usual dose
In all situations, transfer to oral therapy 12 to 24 hours after a loading dose or as soon as practical. See Precautions and Monitor.
Status epilepticus, anticonvulsant:
A loading dose of 10 to 15 mg/kg. Do not exceed a total dose of 1.5 Gm. Lethal dose estimated at 2 to 5 Gm. Another source suggests that 15 to 20 mg/kg is generally recommended. Follow with maintenance doses of 100 mg every 6 to 8 hours. Adjust dose based on phenytoin levels; see Monitor. Other measures, including concomitant administration of an IV benzodiazepine (such as diazepam) or an IV short-acting barbiturate, will usually be necessary for rapid control of seizures because of the required slow rate of administration of phenytoin. If seizure is not terminated, consider other anticonvulsants, barbiturates, or anesthesia.
Pediatric dose
Status epilepticus, anticonvulsant:
15 to 20 mg/kg as a loading dose. Follow with a maintenance dose for age (listed below):
Neonates:
Begin with 5 mg/kg/24 hr in equally divided doses every 12 hours. Range is 5 to 8 mg/kg/24 hr (2.5 to 4 mg/kg every 12 hours).
Infants and other pediatric patients:
Begin with 5 mg/kg/24 hr in equally divided doses every 8 to 12 hours (2.5 mg/kg every 12 hours or 1.67 mg/kg every 8 hours). Range varies according to age:
6 months to 3 years:
8 to 10 mg/kg/24 hr (4 to 5 mg/kg every 12 hours or 2.67 to 3.33 mg/kg every 8 hours).
4 to 6 years:
7.5 to 9 mg/kg/24 hr (3.75 to 4.5 mg/kg every 12 hours or 2.5 to 3 mg/kg every 8 hours).
7 to 9 years:
7 to 8 mg/kg/24 hr (3.5 to 4 mg/kg every 12 hours or 2.3 to 2.6 mg/kg every 8 hours).
10 to 16 years:
6 to 7 mg/kg/24 hr (3 to 3.5 mg/kg every 12 hours or 2 to 2.3 mg/kg every 8 hours).
Dose adjustments
Use caution, lower dose, and slower rate of administration in the seriously ill, elderly, and cachectic patients. ■ Lower doses may also be required in patients with renal or hepatic disease or in those with hypoalbuminemia. Monitoring of unbound (free) phenytoin concentrations may be a better dosing guide. ■ See Drug/Lab Interactions.
Dilution
Available in 100- or 250-mg ampules or vials and in 100-mg syringes. After verifying patency, may be administered directly into a large peripheral or central vein through a large-gauge catheter. Alternately, may be further diluted in NS to a concentration of no less than 5 mg/mL and administered as an infusion. Use solution only when completely dissolved and clear.
Filters:
Manufacturer recommends use of an in-line filter (0.22 to 0.55 microns) when administered as an infusion.
Storage:
Store between 15° and 30° C. Infusion solutions should be administered immediately after preparation and must be completed within 1 to 4 hours. Do not refrigerate infusion solutions.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer recommends not adding to IV solutions other than NS or mixing with other medications and states that “the addition of phenytoin to dextrose or dextrose-containing solutions will result in precipitation.” Always flush line with NS before and after administration of any other drug through the same IV line. See Dilution.
One source suggests the following compatibilities:
Additive:
Not recommended by manufacturer.
Verapamil.
Y-site:
Esmolol (Brevibloc), famotidine (Pepcid IV), fluconazole (Diflucan), tacrolimus (Prograf).
Rate of administration
Because of the risk of local toxicity, IV phenytoin should be administered directly into a large peripheral or central vein through a large-gauge catheter. Before administration, the patency of the IV should be tested with a sterile saline flush. Follow the administration of phenytoin with a saline flush to avoid local venous irritation due to the alkalinity of the solution.
Adults:
Administer slowly at a rate of 25 to 50 mg or fraction thereof over 1 minute. Do not exceed 50 mg/min.
Pediatric patients:
Do not exceed 1 to 3 mg/kg/min. Another source suggests 0.5 mg/kg/min in neonates or 1 mg/kg/min in infants and other pediatric patients not to exceed 50 mg/min.
Elderly:
Limit rate to 25 mg/min.
Infusion:
Should be completed within 1 to 4 hours. Do not exceed 25 to 50 mg/min rate. Best if piggybacked to a compatible primary IV so phenytoin can be discontinued if side effects occur, but IV can be kept open.
Actions
A synthetic anticonvulsant, chemically related to barbiturates. Selectively stabilizes seizure threshold and depresses seizure activity in the motor cortex. Mechanism of action may be due to increasing efflux or decreasing influx of sodium ions across the cell membrane during generation of the action potential. Phenytoin reduces the maximum activity of the brain stem centers responsible for the tonic phase of generalized tonic-clonic seizures. Effective control in emergency treatment of seizures may take 15 to 20 minutes because of rate of injection required. Highly protein bound. Metabolized by cytochrome P450 enzymes CYP2C9 and CYP2C19 and is excreted in changed form in the urine. Half-life ranges from 10 to 15 hours. Crosses the placental barrier. Secreted in breast milk.
Indications and uses
Control of generalized tonic-clonic status epilepticus and prevention and treatment of seizures during neurosurgery. Parenteral phenytoin should be used only when oral phenytoin administration is not possible.
Contraindications
Known hypersensitivity to phenytoin or other hydantoin products. ■ Bradycardia; sinoatrial, second-, or third-degree heart block; Stokes-Adams syndrome. ■ Coadministration with delavirdine (Rescriptor).
Precautions
Discontinue immediately for hypersensitivity reactions; with caution, substitute a nonhydantoin anticonvulsant. When substituting a new anticonvulsant, consideration should be given to avoiding structurally related drugs such as carboxamides (e.g., carbamazepine), barbiturates, succinimides, and oxazolidinediones (e.g., trimethadione). ■ Abrupt withdrawal may cause increased seizure activity. Gradually reduce dose, discontinue, or substitute alternative antiepileptic agents. ■ Not effective for absence seizures; combined therapy is required if both conditions are present. ■ Not indicated for seizures due to hypoglycemia or other metabolic causes. ■ Severe hypotension and cardiac arrhythmias have occurred with rapid infusion. Risk increases with increased rates, but adverse cardiac events have been reported at or below the recommended infusion rates. Careful cardiac monitoring is required during and after IV administration. Because of the risks of cardiac and local toxicity associated with IV phenytoin, oral phenytoin should be used whenever possible. ■ Use caution in hypotension and severe myocardial insufficiency. ■ Use caution with low serum albumin level, and adjust dose as indicated. Phenytoin is highly bound to serum protein (approximately 80% to 90% or more) and a reduced albumin causes an increase in free drug availability. ■ Drug reaction with eosinophilia and systemic symptoms (DRESS), also known as multiorgan hypersensitivity, has been reported. Usually presents with fever, rash, and/or lymphadenopathy in association with other organ system involvement such as hepatitis, nephritis, hematologic abnormalities, myocarditis, or myositis. Eosinophilia is often present. Deaths have been reported. ■ Serious and sometimes fatal dermatologic reactions, including toxic epidermal necrolysis (TEN) and Stevens-Johnson syndrome (SJS), have been reported. Onset of symptoms is usually within 28 days but can occur later. ■ Discontinue phenytoin if skin rash appears unless the rash is clearly not drug related. ■ Selected patients of Asian ancestry (e.g., Han Chinese, Filipino, Malaysian, South Asian Indian, and Thai descent) with a specific human leukocyte antigen allele may have an increased risk of serious skin reactions (e.g., Stevens-Johnson syndrome, toxic epidermal necrolysis) from phenytoin therapy. ■ Cases of acute hepatotoxicity, including hepatic failure, have been reported. These events may be part of the spectrum of DRESS or may occur in isolation. Reactions have included elevated liver function tests, eosinophilia, fever, hepatomegaly, jaundice, leukocytosis, and lymphadenopathy. Discontinue immediately and substitute alternative anticonvulsant therapy. ■ Hematopoietic complications, some fatal, have been reported (e.g., agranulocytosis, granulocytopenia, leukopenia, thrombocytopenia, or pancytopenia with or without bone marrow suppression). ■ Some reports suggest a relationship between phenytoin and the development of lymphadenopathy (local or generalized). Lymph node involvement may occur with or without S/S resembling DRESS. In all cases of lymphadenopathy, follow-up observation for an extended period is indicated, and alternative anticonvulsant therapy should be strongly considered. ■ Local toxicity, including purple glove syndrome (edema, discoloration, and pain distal to the injection site), has occurred and may or may not be associated with extravasation. Irritation may range from slight tenderness to extensive necrosis and sloughing. ■ Inhibits insulin release and may increase serum glucose; monitoring indicated in patients with diabetes. ■ Use with caution in patients with porphyria. Phenytoin may exacerbate this disease. ■ Antiepileptic drugs (AEDs) increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior. Some psychotic symptoms and/or behavioral changes resolved without intervention. Others required dose reduction or discontinuation of the antiepileptic agent.
Monitor:
Narrow margin of error between therapeutic and toxic dose. Plasma levels above 10 mcg/mL usually control seizure activity. The acceptable range is 5 to 20 mcg/mL. Consider monitoring free phenytoin levels in patients with hypoalbuminemia or renal or hepatic insufficiency (therapeutic range is 1 to 2 mcg/mL). Toxicity begins with nystagmus and may be seen at levels less than 20 mcg/mL. Serum levels sustained above the optimum range may produce confusional states referred to as delirium, psychosis, encephalopathy, or rarely irreversible cerebellar dysfunction. ■ Observe patient closely for signs of CNS side effects. ■ Periodic monitoring of CBC, platelets, albumin, urinalysis, and hepatic and renal function is recommended. ■ Monitor ECG and BP continuously. ■ Closely monitor patients who are gravely ill, have impaired liver function, or are elderly. May show early signs of toxicity. ■ Observation of patient symptoms and effectiveness of all medications is imperative. ■ Observe for rash and discontinue if one appears; see Precautions. ■ Determine absolute patency of vein. Avoid extravasation. Very alkaline; follow each injection with sterile NS to reduce local venous irritation. ■ Patients maintained with phenytoin should be given a dose the morning of surgery to maintain adequate serum levels. ■ See Precautions and Drug/Lab Interactions.
Patient education:
May increase the risk of suicidal thoughts and behavior. Promptly report emergence or worsening of S/S of depression, any unusual changes in mood or behavior, or thoughts about self-harm. ■ Women who are pregnant or who become pregnant should be encouraged to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry.
Maternal/child:
Category D: avoid pregnancy. Prenatal exposure to phenytoin may increase the risk for congenital malformations and other adverse development outcomes. Consider risks versus benefit. ■ Alterations in phenytoin kinetics in pregnant women may necessitate periodic monitoring of serum levels. ■ Newborns whose mothers received phenytoin during pregnancy may develop a life-threatening bleeding disorder that can be prevented by giving vitamin K to the mother before delivery and to the neonate after birth. ■ Discontinue breast-feeding.
Elderly:
See Dose Adjustments and Rate of Administration. ■ Clearance tends to decrease with increasing age. ■ Low serum albumin causing a decrease in protein binding may result in increased sensitivity to phenytoin.
Drug/lab interactions
Interactions are numerous and potentially life threatening. Review of drug profile by pharmacist imperative. ■ Coadministration with delavirdine (Rescriptor) is contraindicated. Has potential for loss of virologic response and possible resistance to delavirdine or to the class of nonnucleoside reverse transcriptase inhibitors. ■ Serum levels may be increased by alcohol (acute ingestion), amiodarone (Nexterone), antiepileptic agents (e.g., ethosuximide [Zarontin], felbamate [Felbatol], methsuximide [Celontin], oxcarbazepine [Trileptal], topiramate [Topamax]), azoles (e.g., fluconazole [Diflucan], itraconazole [Sporanox], ketoconazole [Nizoral], miconazole [Oravig], voriconazole [VFEND]), capecitabine (Xeloda), chloramphenicol (Chloromycetin), chlordiazepoxide (Librium), disulfiram (Antabuse), estrogens, fluorouracil (5-FU), fluoxetine (Prozac), fluvastatin (Lescol), fluvoxamine (Luvox), H2 antagonists (e.g., cimetidine [Tagamet]), halothane, isoniazid (INH), methylphenidate (Ritalin), omeprazole (Prilosec), phenothiazines (e.g., prochlorperazine [Compazine]), salicylates (aspirin), sertraline (Zoloft), succinimides, sulfonamides (e.g., sulfadiazine, sulfamethoxazole/trimethoprim [Bactrim], sulfaphenazole), ticlopidine (Ticlid), tolbutamide (Orinase), trazodone (Desyrel), and warfarin (Coumadin). ■ Serum levels and effectiveness may be decreased by antineoplastics usually in combination (e.g., bleomycin [Blenoxane], carboplatin [Paraplatin], cisplatin, doxorubicin [Adriamycin], methotrexate), carbamazepine (Tegretol), chronic alcohol abuse, diazepam (Valium), diazoxide (Proglycem), folic acid, fosamprenavir (Lexiva), nelfinavir (Viracept), reserpine, rifampin (Rifadin), ritonavir (Norvir), St. John’s wort, theophylline, and vigabatrin (Sabril). ■ Phenytoin serum levels may be increased or decreased by phenobarbital (Luminal), valproate sodium (Depacon), and valproic acid (Depakene). Similarly, phenytoin may unpredictably affect the levels and efficacy of these drugs. ■ The addition of withdrawal of drugs while patients are undergoing phenytoin therapy may require an adjustment of the phenytoin dose. ■ Phenytoin will inhibit the effects of azoles (e.g., fluconazole, itraconazole, ketoconazole, posaconazole [Noxafil], voriconazole), corticosteroids, doxycycline, estrogens, furosemide (Lasix), irinotecan (Camptosar), oral contraceptives, paclitaxel (Taxol), paroxetine (Paxil), quinidine, rifampin (Rifadin), sertraline (Zoloft), teniposide (Vumon), theophylline, and vitamin D. Dose adjustment of these agents may be indicated. ■ Phenytoin decreases plasma concentrations of active metabolites of albendazole (Albenza), HIV antivirals (e.g., efavirenz [Sustiva], lopinavir/ritonavir [Kaletra], indinavir [Crixivan], nelfinavir [Viracept], ritonavir [Norvir], saquinavir [Invirase]), antiepileptic agents (e.g., carbamazepine [Tegretol], felbamate [Felbatol], lamotrigine [Lamictal], oxcarbazepine [Trileptal], quetiapine [Seroquel], topiramate [Topomax]), atorvastatin (Lipitor), chlorpropamide (Diabinese), clozapine (Clozaril), cyclosporine (Sandimmune), digoxin (Lanoxin), fluvastatin (Lescol), folic acid, methadone (Dolophine), mexiletine, nifedipine (Procardia), nimodipine, nisoldipine (Sular), praziquantel (Biltricide), simvastatin (Zocor), and verapamil. Adjust doses of these agents as indicated. ■ Phenytoin when given with fosamprenavir alone may decrease the concentration of amprenavir (Agenerase), the active metabolite. Phenytoin when given with the combination of fosamprenavir and ritonavir may increase the concentration of amprenavir (Agenerase). ■ May increase or decrease PT/INR responses when coadministered with warfarin (Coumadin). ■ Chronically administered phenytoin may cause resistance to the neuromuscular blocking action of nondepolarizing neuromuscular blocking agents (e.g., cisatracurium [Nimbex], pancuronium, rocuronium [Zemuron], and vecuronium). Monitor patients closely; recovery from neuromuscular blockade may be more rapid than expected, and infusion rate requirements may be higher. ■ Alters some clinical laboratory tests (e.g., may decrease T4; may increase glucose, alkaline phosphatase, and GGT; and may produce low results in dexamethasone or metyrapone tests).
Side effects
Altered tatste sensation, ataxia, confusion, constipation, decreased coordination, dizziness, drowsiness, dyskinesias, fever, headache, hyperplasia of gums, insomnia, nausea, nervousness, nystagmus, paresthesia, skin eruptions, slurred speech, somnolence, tremors, vertigo, visual disturbances, vomiting.
Major:
Acute hepatic failure, bradycardia, cardiac arrest, cardiovascular collapse, CNS depression, dermatologic reactions (including local toxicity, Stevens-Johnson syndrome, and toxic epidermal necrolysis), DRESS, heart block, hematopoietic complications, hypotension, lymphadenopathy, Peyronie’s disease, respiratory arrest, tonic seizures, toxic hepatitis, ventricular fibrillation. Hypersensitivity reactions, including anaphylaxis, have been reported (rare). Psychotic symptoms, including aggression, agitation, anger, anxiety, apathy, depersonalization, depression, emotional lability, hallucinations, hostility, irritability, and suicidal tendencies, have occurred with antiepileptic agents.
Overdose:
Ataxia, blurred vision, dysarthria, hyperreflexia, lethargy, nausea, nystagmus, slurred speech, tremor, and vomiting.
Antidote
Notify the physician of any side effects. If minor symptoms progress or any major side effect occurs, discontinue the drug and notify the physician; see Precautions. Maintain a patent airway and resuscitate as necessary. Symptoms of heart block or bradycardia may be reversed with IV atropine. Epinephrine may also be useful. Decrease rate or discontinue infusion for severe hypotension or cardiac arrhythmias. Hemodialysis may be useful in overdose.
Phosphate
(FOS-fayt)
Potassium Phosphate, Sodium Phosphate
Electrolyte replenisher
Antihypophosphatemic
pH 5 to 7.8
Usual dose
Dependent on individual needs of the patient.
TPN, adults and pediatric patients:
10 to 15 mM (310 to 465 mg) of phosphorus/liter of TPN solution should maintain normal serum phosphate. Larger amounts may be required. 1 mM equals 31 mg.
Acute hypophosphatemia: Adults and pediatric patients:
0.08 to 0.32 mM/kg of body weight as a loading dose equally distributed over 6 hours. Maintain pediatric patients with 0.5 to 1.5 mM/kg/24 hr. Maintain adults with 48.4 to 64.5 mM/24 hr.
Infant dose
Infants receiving TPN:
1.5 to 2 mM/kg of body weight/day.
Dose adjustments
Lower-end initial doses may be indicated in the elderly based on the potential for decreased organ function and concomitant disease or drug therapy.
Dilution
Must be diluted in a larger volume of suitable IV solution and given as an infusion. Soluble in most commonly used IV solutions (see chart on inside back cover) except protein hydrolysate. Mix thoroughly. See Compatibility.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
All formulations
Mix thoroughly after each addition of supposedly compatible drugs or solutions. TPN solutions requiring the addition of phosphates and calcium salts must be mixed by the pharmacist to avoid a precipitate of calcium phosphate. Specific amounts, calculations, order, and temperature (precipitate forms more readily at room temperature) are required. Deaths have been reported.
One source suggests the following compatibilities:
Potassium phosphate
Additive:
See comments under all formulations.
Mix thoroughly. Metoclopramide (Reglan), verapamil.
Y-site:
Diltiazem (Cardizem), enalaprilat (Vasotec IV), esmolol (Brevibloc), famotidine (Pepcid IV), 6% hydroxyethyl starch (Voluven), labetalol, micafungin (Mycamine), nicardipine (Cardene IV), nitroprusside sodium, telavancin (Vibativ).
Sodium phosphate
Y-site:
Doripenem (Doribax), micafungin (Mycamine), telavancin (Vibativ).
Rate of administration
A usual dose is usually equally distributed over 6 hours. Other sources suggest administering up to 15 mM over 2 hours, up to 30 mM over 4 hours, and up to 45 mM over 6 hours. Potassium phosphate will be further limited by the maximum rate for potassium. Consider sodium/potassium content. Infuse slowly. Rapid infusion may cause phosphate or potassium intoxication. Serum calcium may be reduced rapidly, causing hypocalcemic tetany.
Actions
Involved in bone deposition. Helps to maintain calcium levels, has a buffering effect on acid-base equilibrium, and influences renal excretion of the hydrogen ion. Normal levels in adults, 3 to 4.5 mg/dL of serum; in pediatric patients, 4 to 7 mg/dL. Excreted in urine.
Indications and uses
To prevent or correct hypophosphatemia in patients with restricted or no oral intake.
Contraindications
Any disease with high phosphate or low calcium levels, hyperkalemia (potassium phosphate), hypernatremia (sodium phosphate).
Precautions
Rapid infusion may cause phosphate, sodium, or potassium intoxication. Serum calcium may be reduced, rapidly causing hypocalcemic tetany. ■ Use sodium phosphate with caution in renal impairment, cirrhosis, cardiac failure, or any edematous, sodium-retaining state. ■ Use potassium phosphate with caution in cardiac disease, renal disease, and digitalized patients. ■ See Compatibility.
Monitor:
Monitor serum calcium, potassium, phosphate, chlorides, and sodium. Discontinue when serum phosphate exceeds 2 mg/dL. ■ See Drug/Lab Interactions.
Maternal/child:
Category C: safety for use in pregnancy not established.
Elderly:
Differences in response between elderly and younger patients have not been identified. Lower-end initial doses may be appropriate in the elderly; see Dose Adjustments.
Drug/lab interactions
May cause hyperkalemia with potassium-sparing diuretics (e.g., amiloride) or angiotensin-converting enzyme inhibitors (e.g., enalapril [Vasotec]).
Side effects
Elevated phosphates, reduced calcium levels and hypocalcemic tetany, elevated potassium levels causing cardiac arrhythmias, flaccid paralysis, heaviness of the legs, hypotension, listlessness, mental confusion, paresthesia of the extremities.
Antidote
For any side effect, discontinue the drug and notify the physician. Restore serum calcium with calcium gluconate or chloride. Shift potassium from serum to cells with 150 mL of 1/6 M sodium lactate or 10% to 20% dextrose with 10 units regular insulin for each 20 Gm dextrose at 300 to 500 mL/hr. Correct acidosis with sodium bicarbonate. Reduce sodium by restriction, diuretics, or hemodialysis. Resuscitate as necessary.
Physostigmine salicylate
(fye-zoh-STIG-meen sah-LIS-ah-layt)
Cholinergic
Cholinesterase inhibitor
Antidote
pH 5.8
Usual dose
Postanesthesia:
0.5 to 1 mg initially. Repeat at 10- to 30-minute intervals until desired results obtained.
Anticholinergic toxicity:
0.5 to 2 mg initially. 1 to 4 mg may be repeated as necessary as life-threatening signs recur (arrhythmias, convulsions, deep coma). Maximum dose is 4 mg in 30 minutes.
Pediatric dose
To be used in life-threatening situations only. 0.02 mg/kg/dose. May be repeated at 5- to 10-minute intervals only if toxic effects persist and there is no sign of cholinergic effects. Maximum total dose is 2 mg. See Maternal/Child.
Dilution
May be given undiluted. Do not add to IV solutions. May be given through Y-tube or three-way stopcock of infusion set.
Compatibility
Specific information not available. Consider specific use; consult pharmacist.
Rate of administration
Rapid IV administration may cause bradycardia, hypersalivation, respiratory distress, and convulsions.
1 mg or fraction thereof over 1 to 3 minutes.
Pediatric rate:
0.5 mg or fraction thereof over at least 1 minute.
Actions
An extract of Physostigma venenosum seeds. It inhibits the destructive action of acetylcholinesterase and prolongs and exaggerates the effects of acetylcholine. Stimulates parasympathetic nerve stimulation (pupil contraction, increased intestinal musculature tonus, bronchial constriction, salivary and sweat gland stimulation). Does enter the CNS. Onset of action occurs in 5 minutes and lasts about 1 hour. Rapidly hydrolyzed by cholinesterases.
Indications and uses
To reverse CNS toxic effects caused by drugs capable of producing anticholinergic poisoning (e.g., atropine), other anticholinergic/antispasmodic agents (e.g., phenothiazines, antihistamines), anticholinergic antiparkinson agents (e.g., benztropine [Cogentin], trihexyphenidyl [Artane]), and tricyclic antidepressants (e.g., imipramine [Tofranil]).
Unlabeled uses:
Treatment of delirium tremens.
Contraindications
Asthma, cardiovascular disease, diabetes, gangrene, mechanical obstruction of the intestines or urogenital tract, vagotonic states, patients receiving choline esters, depolarizing neuromuscular blocking agents (succinylcholine), or tricyclic antidepressants (e.g., amitriptyline [Elavil]).
Precautions
Rapid IV administration may cause bradycardia, hypersalivation, respiratory distress, and convulsions. ■ Contains bisulfites; use caution in allergic individuals.
Monitor:
Atropine must always be available. ■ Monitor vital signs. ■ See Drug/Lab Interactions.
Maternal/child:
Safety for use in pregnancy and breast-feeding not established. ■ Has caused muscular weakness in neonates of mothers treated with other cholinesterase inhibitors for myasthenia gravis. ■ May contain benzyl alcohol; do not use in neonates.
Drug/lab interactions
Potentiates succinylcholine and other choline esters (e.g., bethanecol). ■ May antagonize CNS depressant effects of diazepam (Valium). ■ May cause serious complications, including death, with tricyclic antidepressants (e.g., amitriptyline [Elavil]).
Side effects
Anxiety, bradycardia, cholinergic crisis (overdose), coma, convulsions, defecation, delirium, disorientation, emesis, hallucinations, hyperactivity, hypersalivation, hypersensitivity, nausea, respiratory distress, salivation, seizures, sweating, urination.
Antidote
Keep physician informed of side effects. For excessive nausea or sweating, reduce dose. Discontinue drug for bradycardia; convulsions; excessive defecation, emesis, salivation, or urination; or respiratory distress. Treat cholinergic side effects (e.g., arrhythmias, bronchoconstriction) or hypersensitivity with the specific antagonist atropine sulfate in doses of 0.6 mg IV. May be repeated every 3 to 10 minutes. Endotracheal intubation or tracheostomy are considered prophylactic in anesthesia or crisis. Artificial ventilation, oxygen therapy, cardiac monitoring, adequate suctioning, and treatment of shock or convulsions must be instituted and maintained as necessary.
Phytonadione 
(fye-toe-nah-DYE-ohn)
Vitamin K1
Vitamin (prothrombinogenic)
Antidote
Antihemorrhagic
pH 5 to 7
Usual dose
Should be given by the SC or oral route whenever possible; parenteral route administration has caused death; see Precautions. A single dose is preferred, but it may be repeated if clinically indicated.
Vitamin K deficiency:
Up to 10 mg may be added to TPN solutions as indicated.
Anticoagulant-induced (warfarin or dicumarol) hypoprothrombinemia:
2.5 to 10 mg. Doses up to 25 mg and, rarely, 50 mg may be needed. May repeat in 6 to 8 hours if initial response is not adequate. Doses as low as 1 to 2 mg may be effective. Use the smallest dose that achieves effective results to prevent clotting hazards.
Hypoprothrombinemia from other causes:
2 to 25 mg (rarely 50 mg), depending on the severity of the deficiency and the response obtained.
Pediatric dose
See Usual Dose, Precautions, and Maternal/Child.
Vitamin K deficiency:
1 to 2 mg may be added to TPN solutions as indicated.
Anticoagulant-induced (warfarin or dicumarol) hypoprothrombinemia in infants and children:
1 to 2 mg/dose.
Hypoprothrombinemia from other causes in infants and children:
A single dose of 1 to 2 mg.
Newborn dose
See Usual Dose, Precautions, and Maternal/Child. Rarely given IV in the newborn. SC injection preferred.
Prophylaxis of hemorrhagic disease of the newborn:
0.5 to 1 mg IM within 1 hour of birth.
Treatment of hemorrhagic disease of the newborn:
1 to 2 mg/24 hr SC or IM. Higher doses may be necessary if the mother has been receiving oral anticoagulants (e.g., warfarin [Coumadin]) or anticonvulsants (e.g., phenytoin [Dilantin]). Whole blood or blood components may be indicated for excessive bleeding. Give phytonadione concurrently to correct the underlying disorder.
Dose adjustments
Dilution
Use only preservative-free solutions. May be diluted only with NS, D5NS, or D5W. Dilution with at least 10 mL of diluent is recommended to facilitate prescribed rate of administration. Photosensitive; protect from light in all dilutions. Use immediately after preparation.
Filters:
Not required by manufacturer; however, there should be no significant loss of potency with the use of a 0.22-micron filter.
Storage:
Photosensitive; protect from light before use and in all dilutions. Store unopened ampules below 40° C (104° F), preferably between 15° C and 30° C (59° F and 86° F). Protect from light and freezing. Discard diluted solution and drug remaining in ampule after single use.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Amikacin, chloramphenicol (Chloromycetin), sodium bicarbonate.
Y-site:
Ampicillin, epinephrine (Adrenalin), famotidine (Pepcid IV), heparin, hydrocortisone sodium succinate (Solu-Cortef), potassium chloride (KCl).
Rate of administration
Each 1 mg or fraction thereof over 1 minute or longer. Too-rapid injection has caused severe reactions, including fatalities.
Actions
Vitamin K, a fat-soluble vitamin, is essential for hepatic production of four blood coagulation factors including prothrombin. These are required for normal blood clotting. Onset of action is within 1 to 2 hours. Usually controls hemorrhage in 3 to 6 hours; normal prothrombin levels should be obtained in 12 to 14 hours. Metabolized by the liver and eliminated in urine and bile.
Indications and uses
Coagulation disorders due to faulty formation of factors II, VII, IX, and X when caused by vitamin K deficiency or interference with vitamin K activity. Indicated in anticoagulant-induced prothrombin deficiency (warfarin or dicumarol). ■ Prophylaxis and treatment of hemorrhagic disease of the newborn. ■ Hypoprothrombinemia resulting from antibacterial therapy. ■ Hypoprothrombinemia secondary to factors limiting the absorption or synthesis of vitamin K (e.g., obstructive jaundice, biliary fistula, sprue, ulcerative colitis, celiac disease, intestinal resection, cystic fibrosis of the pancreas, and regional enteritis). ■ Other drug-induced hypoprothrombinemia where it is definitely shown that the result is due to interference with vitamin K metabolism (e.g., salicylates).
Contraindications
Hypersensitivity to components.
Precautions
IV and/or IM is not the route of choice; used only when SC or oral route cannot be used. Use extreme caution; has caused severe reactions, including death, with the first injection, even when the product has been diluted and infused slowly. ■ As an alternative to administering phytonadione, discontinuation or reduction of the doses of drugs interfering with coagulation mechanisms (e.g., antibiotics, salicylates) should be considered. The severity of the coagulation disorder should determine if phytonadione is required in addition to discontinuing or reducing the doses of interfering drugs. To correct excess anticoagulation after the use of warfarin (Coumadin) (e.g., returning an increased INR to the desired range), consider the degree of elevation and the presence of clinically significant bleeding. ■ Supplement with whole blood transfusion or blood components if indicated. ■ Now the only vitamin K product for IV use. ■ Do not use to counteract anticoagulant effects of heparin; not effective. Protamine sulfate is indicated. ■ Does not restore abnormal platelet function to normal. ■ Does not correct hypoprothrombinemia due to hepatocellular damage. ■ When phytonadione is used in a patient for whom anticoagulant therapy is indicated, the same clotting hazards that existed before beginning anticoagulant therapy will recur. Use the smallest dose of phytonadione possible and monitor PT.
Monitor:
See Neonatal Dose, Precautions, and Drug/Lab Interactions. ■ Dose and effect determined by PT/INR. Repeat PT/INR 6 to 8 hours after a dose. Keep the physician informed. ■ Pain and swelling at injection site can occur.
Maternal/child:
Category C: safety for use not established. ■ Use caution during breast-feeding. ■ Use extreme caution in premature infants and neonates. Hemolysis, jaundice, and hyperbilirubinemia in newborns may be related to the dose of phytonadione. The recommended dose should not be exceeded. Severe hemolytic anemia, hemoglobinuria, kernicterus, brain damage, and death may occur. ■ Neonates with hemorrhagic disease of the newborn should respond to administration of phytonadione with a shortening of the PT within 2 to 4 hours. If shortening of the PT is not seen, consider other coagulation disorders. ■ May contain benzyl alcohol. When used as recommended, there is no evidence to suggest that this small amount is associated with toxicity.
Drug/lab interactions
Discontinue drugs adversely affecting the coagulation mechanism if possible (e.g., salicylates, antibiotics). ■ May cause temporary resistance to prothrombin-depressing oral anticoagulants by increasing amount of phytonadione in the liver and blood. Anticoagulation will require larger doses of same or use of heparin sodium.
Side effects
Cyanosis, diaphoresis, dizziness, dyspnea, hyperbilirubinemia, hypotension, injection site reactions, peculiar taste sensations, rapid and weak pulse, tachycardia, transient flushing sensation. Anaphylaxis, cardiac and/or respiratory arrest, shock, and death have occurred with parenteral route.
Antidote
Should not be necessary if dosage is accurately calculated before administration. Action can be reversed by warfarin or heparin if indicated. Discontinue the drug and notify the physician of any side effects. For most side effects the physician will probably choose to continue the drug at a decreased rate of administration. Treat hypersensitivity reactions as necessary.
Piperacillin sodium and tazobactam sodium
(pie-PER-ah-sill-in SO-dee-um and tay-zoh-BAC-tam SO-dee-um)
Zosyn
Antibacterial (extended-spectrum penicillin and beta-lactamase inhibitor)
Usual dose
Measurement of both drugs is included in the total dose; for every 1 Gm of piperacillin there is 0.125 Gm of tazobactam (8:1 ratio).
12 Gm piperacillin/1.5 Gm tazobactam/24 hours given as 3.375 (3 Gm piperacillin/0.375 Gm tazobactam) every 6 hours. Usual duration of therapy is 7 to 10 days, based on severity of infection and patient progress.
Nosocomial pneumonia:
4.5 Gm every 6 hours. Also use an aminoglycoside. Continue aminoglycoside if P. aeruginosa is isolated; may be discontinued if it is not isolated. Usual duration of therapy is 7 to 14 days.
Pediatric dose
2 months to 9 months of age:
80 mg piperacillin/10 mg tazobactam per kg of body weight every 8 hours.
Over 9 months of age weighing up to 40 kg:
100 mg piperacillin/12.5 mg tazobactam per kg of body weight every 8 hours.
Pediatric patients over 40 kg:
See Usual Dose.
Dose adjustments
No dose adjustment needed in impaired liver function. ■ Dose reduction may be required in the elderly. ■ Adjust dose in adult patients with impaired renal function based on the following chart. ■ Dose recommendations for pediatric patients with impaired renal function are not available.
| Piperacillin/Tazobactam Dose Guidelines in Adults with Impaired Renal Function | ||
| Renal Function (Creatinine Clearance, mL/min) | All Indications (Except Nosocomial Pneumonia) | Nosocomial Pneumonia |
| >40 mL/min | 3.375 Gm q 6 hr | 4.5 Gm q 6 hr |
| 20-40 mL/min* | 2.25 Gm q 6 hr | 3.375 Gm q 6 hr |
| <20 mL/min* | 2.25 Gm q 8 hr | 2.25 Gm q 6 hr |
| Hemodialysis† | 2.25 Gm q 12 hr | 2.25 Gm q 8 hr |
| CAPD | 2.25 Gm q 12 hr | 2.25 Gm q 8 hr |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


