(PACK-lih-tax-el)
Onxol, Taxol ■ Abraxane
Antineoplastic (taxane)
pH 4.4 to 5.6 ■ not available
Usual dose
Assessment required before dosing: see Precautions, Monitor, and Dose Adjustments.
Conventional paclitaxel
Several regimens of paclitaxel, alone or in combination with other antineoplastics, are in use. Doses vary, depending on the regimen used. Consult literature. ■ For all uses, premedication, specific parameters, and specific equipment are required before or during administration; see Premedication, Dose Adjustments, and Precautions/Monitor.
Premedication:
Must be premedicated before each dose to prevent severe hypersensitivity reactions. Usual regimen includes oral dexamethasone (Decadron) 20 mg 12 and 6 hours before; IV diphenhydramine (Benadryl) 50 mg 30 to 60 minutes before; and an H2 antagonist (e.g., ranitidine [Zantac] 50 mg or famotidine [Pepcid IV] 20 mg) 30 to 60 minutes before dosing with paclitaxel. When premedicating patients with AIDS-related Kaposi’s sarcoma, reduce the dose of dexamethasone to 10 mg at 12 and 6 hours before paclitaxel. The doses of IV diphenhydramine and IV H2 antagonists remain as above.
Ovarian cancer in previously untreated patients:
135 mg/M2 as an infusion over 24 hours. Follow with cisplatin 75 mg/M2 as an infusion over 6 to 8 hours. Repeat every 3 weeks. An alternative regimen is paclitaxel 175 mg/M2 as an infusion over 3 hours. Follow with cisplatin 75 mg/M2 (one source suggests an infusion over 24 hours; another suggests 6 to 8 hours, which would allow for outpatient therapy). Repeat every 3 weeks. See comments under Usual Dose.
Ovarian cancer in patients previously treated with chemotherapy:
135 or 175 mg/M2 as an infusion over 3 hours. An alternate regimen suggests the same dose given as a 24-hour infusion. Repeat every 3 weeks. Larger doses, with or without filgrastim (G-CSF, Neupogen), have produced similar responses. See comments under Usual Dose.
Adjuvant treatment of node-positive breast cancer:
175 mg/M2 as an infusion over 3 hours. Repeat every 3 weeks for four courses. Administered sequentially to doxorubicin-containing combination therapy. Clinical trials used four courses of doxorubicin and cyclophosphamide. Administer filgrastim (G-CSF) 5 mcg/kg/dose on Days 3 through 10. See comments under Usual Dose.
Breast cancer in patients previously treated with chemotherapy:
175 mg/M2 as an infusion over 3 hours. Repeat every 3 weeks. An alternate regimen suggests 175 to 250 mg/M2 over 3 hours every 3 weeks. See comments under Usual Dose.
First-line treatment of non–small-cell lung cancer:
135 mg/M2 as an infusion over 24 hours. Follow with cisplatin 75 mg/M2 over 6 to 8 hours. Repeat every 3 weeks. See comments under Usual Dose. A Canadian source recommends 175 mg/M2 as an infusion over 3 hours followed with cisplatin 75 mg/M2 over 6 to 8 hours and repeated every 3 weeks.
AIDS-related Kaposi’s sarcoma:
135 mg/M2 as an infusion over 3 hours. An alternate regimen suggests the same dose given as a 24-hour infusion. Repeat every 3 weeks. Another regimen is 100 mg/M2 as an infusion over 3 hours repeated every 2 weeks. Toxicity somewhat increased with 135 mg/M2 dose in clinical studies. See comments under Usual Dose.
Abraxane
Premedication:
Generally not required; see Precautions.
Metastatic breast cancer (MBC):
260 mg/M2 as an infusion over 30 minutes every 3 weeks.
Non–small-cell lung cancer (NSCLC):
100 mg/M2 as an infusion over 30 minutes on Days 1, 8, and 15 of each 21-day cycle. Given in combination with carboplatin on Day 1 only of each 21-day cycle, beginning immediately after the completion of Abraxane administration.
Adenocarcinoma of the pancreas:
125 mg/M2 as an infusion over 30 to 40 minutes followed by gemcitabine 1,000 mg/M2 as an infusion over 30 to 40 minutes on Days 1, 8, and 15 of each 28-day cycle.
Dose adjustments
Conventional paclitaxel
Reduce dose by 20% for subsequent courses in patients who experience severe peripheral neuropathy or severe neutropenia (neutrophils less than 500 cells/mm3) for 1 week or longer. ■ Withhold therapy if neutrophils below 1,500/mm3 or platelets below 100,000/mm3. ■ Dose reduction not required in impaired renal function. ■ In AIDS-related Kaposi’s sarcoma the parameters are slightly different. Initiate or repeat paclitaxel only if neutrophil count is equal to or greater than 1,000/mm3; reduce dose of dexamethasone to 10 mg/dose; reduce dose of paclitaxel by 20% in patients who experience severe neutropenia (neutrophils less than 500/mm3 for a week or longer); use concomitant filgrastim (G-CSF) as clinically indicated. ■ Recommendations for dose adjustment of the initial course of therapy in patients with impaired hepatic function are listed in the following chart.
| Guidelines for Dose Adjustment of Paclitaxel in Impaired Hepatic Function* | |||
| Degree of Hepatic Impairment | Recommended TAXOL Dose‡ | ||
| Transaminase Levels | Bilirubin Levels† | ||
| 24-Hour Infusion | |||
| <2 × ULN | and | ≤1.5 mg/dL | 135 mg/M2 |
| 2 to <10 × ULN | and | ≤1.5 mg/dL | 100 mg/M2 |
| <10 × ULN | and | 1.6-7.5 mg/dL | 50 mg/M2 |
| ≥10 × ULN | or | >7.5 mg/dL | Not recommended |
| 3-Hour Infusion | |||
| <10 × ULN | and | ≤1.25 × ULN | 175 mg/M2 |
| <10 × ULN | and | 1.26-2 × ULN | 135 mg/M2 |
| <10 × ULN | and | 2.01-5 × ULN | 90 mg/M2 |
| ≥10 × ULN | or | >5 × ULN | Not recommended |
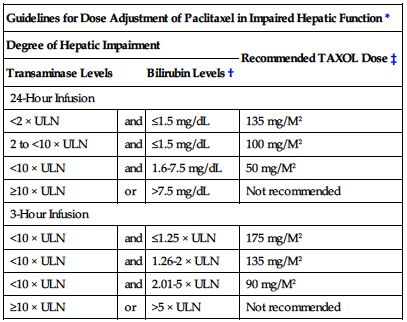
*These recommendations are based on clinical trials of dosages for patients without hepatic impairment of 135 mg/M2 over 24 hours or 175 mg/M2 over 3 hours; data are not available to make dose adjustment recommendations for other regimens (e.g., for AIDS-related Kaposi’s sarcoma).
†Differences in criteria for bilirubin levels between the 3- and 24-hour infusion are due to differences in clinical trial design.
‡Dosage recommendations are for the first course of therapy; further dose reduction in subsequent courses should be based on individual tolerance.
Abraxane
Adjustment of starting dose is not necessary in patients with mild to moderate renal impairment (CrCl equal to or greater than 30 to less than 90 mL/min). ■ Withhold therapy in all patients if neutrophils below 1,500/mm3. ■ In patients with MBC, withhold therapy if platelets below 100,000/mm3. ■ For MBC, reduce dose to 220 mg/M2 in patients who experience severe neutropenia (neutrophils less than 500 cells/mm3 for 7 days or more) or severe sensory neuropathy. Further reduce dose to 180 mg/M2 for subsequent courses if severe neutropenia (less than 500 cells/mm3 for 7 days or more) or severe sensory neuropathy recurs. ■ If Grade 3 sensory neuropathy occurs, withhold treatment until resolution to Grade 1 or 2 for MBC or until resolution to less than or equal to Grade 1 for NSCLC, followed by a dose reduction for all subsequent courses. ■ No dose adjustment is necessary for patients with mild hepatic impairment (total bilirubin greater than ULN and less than or equal to 1.5 times the ULN and AST less than or equal to 10 times the ULN), regardless of indication. ■ Do not administer Abraxane to patients with total bilirubin greater than 5 times the ULN or AST greater than 10 times the ULN regardless of indication because these patients have not been studied. ■ Do not administer Abraxane to patients with metastatic adenocarcinoma of the pancreas who have moderate to severe hepatic impairment. ■ Recommendations for a starting dose in patients with hepatic impairment are shown in the following chart. Doses for subsequent cycles should be based on patient tolerance.
| Abraxane Starting Dose Recommendations for Patients with Hepatic Impairment | ||||||
| Degree of Hepatic Impairment | SGOT (AST) Levels | Bilirubin Levels | Abraxane Dose* | |||
| MBC | NSCLC‡ | Pancreatic‡ Adenocarcinoma | ||||
| Mild | <10 × ULN | and | >ULN to ≤1.5 × ULN | 260 mg/M2 | 100 mg/M2 | 125 mg/M2 |
| Moderate | <10 × ULN | and | >1.5 to ≤3 × ULN | 200 mg/M2† | 80 mg/M2† | N/R |
| Severe | <10 × ULN | and | >3 to ≤5 × ULN | 200 mg/M2† | 80 mg/M2† | N/R |
| >10 × ULN | or | >5 × ULN | N/R | N/R | N/R | |
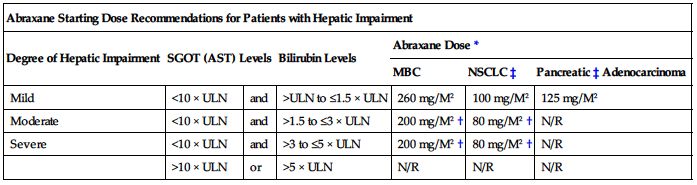
*Dose recommendations are for the first course of therapy. The need for further dose adjustments in subsequent courses should be based on individual tolerance.
†A dose increase to 260 mg/M2 for patients with MBC or to 100 mg/M2 for patients with NSCLC in subsequent courses should be considered if the patient tolerates the reduced dose for two cycles.
‡Patients with bilirubin levels above the ULN were excluded from clinical trials for pancreatic or lung cancer.
MBC, Metastatic breast cancer; N/R, Not recommended; NSCLC, non–small-cell lung cancer.
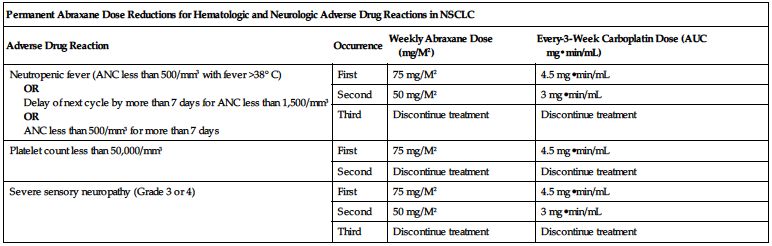
■ In patients with NSCLC who develop severe neutropenia or thrombocytopenia, withhold therapy until counts recover to an ANC of at least 1,500 cells/mm3 and platelets of at least 100,000 cells/mm3 on Day 1 or to an ANC of at least 500 cells/mm3 and platelets of at least 50,000 cells/mm3 on Day 8 or 15 of the cycle. Upon resumption of dosing, follow dose reduction outlined in the following chart.
| Permanent Abraxane Dose Reductions for Hematologic and Neurologic Adverse Drug Reactions in NSCLC | |||
| Adverse Drug Reaction | Occurrence | Weekly Abraxane Dose (mg/M2) | Every-3-Week Carboplatin Dose (AUC mg•min/mL) |
| Neutropenic fever (ANC less than 500/mm3 with fever >38° C) OR Delay of next cycle by more than 7 days for ANC less than 1,500/mm3 OR ANC less than 500/mm3 for more than 7 days | First | 75 mg/M2 | 4.5 mg•min/mL |
| Second | 50 mg/M2 | 3 mg•min/mL | |
| Third | Discontinue treatment | Discontinue treatment | |
| Platelet count less than 50,000/mm3 | First | 75 mg/M2 | 4.5 mg•min/mL |
| Second | Discontinue treatment | Discontinue treatment | |
| Severe sensory neuropathy (Grade 3 or 4) | First | 75 mg/M2 | 4.5 mg•min/mL |
| Second | 50 mg/M2 | 3 mg•min/mL | |
| Third | Discontinue treatment | Discontinue treatment | |
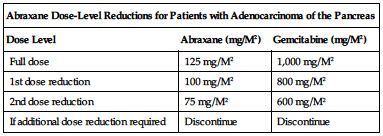
■ Dose-level reductions for patients with adenocarcinoma of the pancreas are outlined in the following chart.
| Abraxane Dose-Level Reductions for Patients with Adenocarcinoma of the Pancreas | ||
| Dose Level | Abraxane (mg/M2) | Gemcitabine (mg/M2) |
| Full dose | 125 mg/M2 | 1,000 mg/M2 |
| 1st dose reduction | 100 mg/M2 | 800 mg/M2 |
| 2nd dose reduction | 75 mg/M2 | 600 mg/M2 |
| If additional dose reduction required | Discontinue | Discontinue |
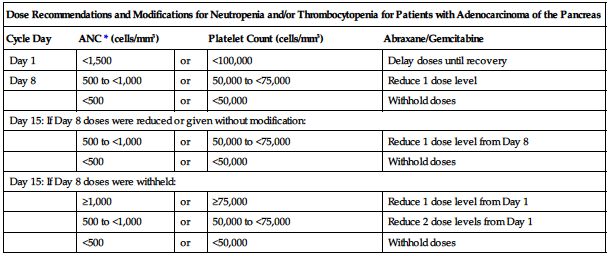
■ Dose modifications for neutropenia and/or thrombocytopenia for patients with adenocarcinoma of the pancreas are provided in the following chart.
| Dose Recommendations and Modifications for Neutropenia and/or Thrombocytopenia for Patients with Adenocarcinoma of the Pancreas | ||||
| Cycle Day | ANC* (cells/mm3) | Platelet Count (cells/mm3) | Abraxane/Gemcitabine | |
| Day 1 | <1,500 | or | <100,000 | Delay doses until recovery |
| Day 8 | 500 to <1,000 | or | 50,000 to <75,000 | Reduce 1 dose level |
| <500 | or | <50,000 | Withhold doses | |
| Day 15: If Day 8 doses were reduced or given without modification: | ||||
| 500 to <1,000 | or | 50,000 to <75,000 | Reduce 1 dose level from Day 8 | |
| <500 | or | <50,000 | Withhold doses | |
| Day 15: If Day 8 doses were withheld: | ||||
| ≥1,000 | or | ≥75,000 | Reduce 1 dose level from Day 1 | |
| 500 to <1,000 | or | 50,000 to <75,000 | Reduce 2 dose levels from Day 1 | |
| <500 | or | <50,000 | Withhold doses | |
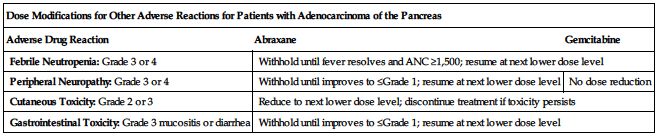
■ Dose modifications for other adverse reactions for patients with adenocarcinoma of the pancreas are provided in the following chart.
| Dose Modifications for Other Adverse Reactions for Patients with Adenocarcinoma of the Pancreas | ||
| Adverse Drug Reaction | Abraxane | Gemcitabine |
| Febrile Neutropenia: Grade 3 or 4 | Withhold until fever resolves and ANC ≥1,500; resume at next lower dose level | |
| Peripheral Neuropathy: Grade 3 or 4 | Withhold until improves to ≤Grade 1; resume at next lower dose level | No dose reduction |
| Cutaneous Toxicity: Grade 2 or 3 | Reduce to next lower dose level; discontinue treatment if toxicity persists | |
| Gastrointestinal Toxicity: Grade 3 mucositis or diarrhea | Withhold until improves to ≤Grade 1; resume at next lower dose level | |
Dilution
Specific techniques required; see Precautions.
Conventional paclitaxel
Must be diluted and given as an infusion. May leach the toxic plasticizer DEHP from PVC infusion bags or sets; prepare and store in bottles (glass, polypropylene) or plastic bags (polypropylene, polyolefin) and administer through polyethylene-lined administration sets. Compatible with NS, D5W, D5NS, or D5R. Final concentration of 0.3 to 1.2 mg/mL required. For a 135 mg/M2 dose, a large adult (body surface about 2 M2) will receive 270 mg (45 mL of paclitaxel at 6 mg/mL). Will require dilution in an additional 180 mL to make a 1.2 mg/mL concentration or in an additional 855 mL to make a 0.3 mg/mL concentration. Solution may appear hazy. Do not use a chemo-dispensing pin; can cause the stopper to collapse and result in loss of sterility.
Abraxane
Available in single-use vials, each containing 100 mg (5 mg/mL after reconstitution with 20 mL of NS). Calculate the exact number of vials needed to achieve the total dosing volume of suspension required.
Total # of vials required = Total dose (mg) ÷ 100 mg
Dosing volume (mL) = Total dose (mg) ÷ 5 (mg/mL)
For example, a MBC patient with a body surface area (BSA) of 1.73 M2 would need a dose of 449.8 mg of Abraxane. 449.8 mg divided by 100 mg equals 4.498 vials, so 5 vials of Abraxane would be needed. 449.8 mg divided by 5 (mg/mL) equals a dosing volume of 90 mL of reconstituted solution.
Reconstitute each 100-mg vial with 20 mL of NS. A specific process is required to avoid foaming or clumping. Over a minimum of 1 minute, slowly inject the NS, directing it to the inside wall of the vial. Do not allow the NS to flow directly onto the lyophilized cake (will cause foaming). Allow each vial to sit for a minimum of 5 minutes while the NS wets the cake. Gently swirl and/or invert each vial slowly for at least 2 minutes until complete dissolution. Avoid generation of foam. If foaming or clumping occurs, allow solution to stand for at least 15 minutes until foam subsides. Solution should be milky and homogenous without visible particulates. If particulates are visible, gently invert to ensure complete resuspension before use.
Withdraw the calculated volume and inject into an empty sterile polyvinyl chloride (PVC) container or a PVC or non–PVC-type infusion bag (a total dosing volume of 90 mL in the previous example). The use of medical devices containing silicone oil as a lubricant (i.e., syringes and IV bags) to reconstitute and administer Abraxane may result in the formation of proteinaceous strands. Discard suspension if proteinaceous strands, particulate matter, or discoloration are observed.
Filters:
Conventional paclitaxel:
Use of an in-line filter not greater than 0.22 microns required for administration.
Storage:
Conventional paclitaxel:
May be stored at CRT or refrigerated before dilution (may appear precipitated under refrigeration; will redissolve at room temperature). Diluted for infusion, it is stable at room temperature for up to 27 hours.
Abraxane:
Store vials in original package at 20° to 25° C (68° to 77° F). Protect from light. Refrigeration or freezing does not affect stability of the product. Immediate use of reconstituted solution is preferred, but reconstituted vial may be refrigerated at 2° to 8° C (36° to 46° F) for a maximum of 24 hours if necessary. If refrigeration required, return to original carton to protect from light. Ensure complete resuspension after removing from refrigerator by gently inverting. Discard unused portions of the vial. Reconstituted solution in an infusion bag should be used immediately; however, it may be refrigerated and protected from bright light for a maximum of 24 hours. The total combined refrigerated storage time of reconstituted Abraxane in the vial and in the infusion bag is 24 hours. This may be followed by storage in the infusion bag at ambient temperature and lighting conditions for a maximum of 4 hours.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Conventional paclitaxel
Leaches out plasticizers; see Dilution.
Conventional paclitaxel
Additive:
Carboplatin (Paraplatin), cisplatin, doxorubicin (Adriamycin); see Drug/Lab Interactions.
Y-site:
Acyclovir (Zovirax), amikacin, aminophylline, ampicillin/sulbactam (Unasyn), anidulafungin (Eraxis), bleomycin (Blenoxane), butorphanol (Stadol), calcium chloride, carboplatin (Paraplatin), cefotetan, ceftazidime (Fortaz), ceftriaxone (Rocephin), cisplatin, cladribine (Leustatin), cyclophosphamide (Cytoxan), cytarabine (ARA-C), dacarbazine (DTIC), dexamethasone (Decadron), diphenhydramine (Benadryl), doripenem (Doribax), doxorubicin (Adriamycin), droperidol (Inapsine), etoposide (VePesid), etoposide phosphate (Etopophos), famotidine (Pepcid IV), fluconazole (Diflucan), fluorouracil (5-FU), furosemide (Lasix), ganciclovir (Cytovene IV), gemcitabine (Gemzar), gentamicin, granisetron (Kytril), heparin, hydrocortisone sodium succinate (Solu-Cortef), hydromorphone (Dilaudid), ifosfamide (Ifex), linezolid (Zyvox), lorazepam (Ativan), magnesium sulfate, mannitol, meperidine (Demerol), mesna (Mesnex), methotrexate, metoclopramide (Reglan), morphine, nalbuphine, ondansetron (Zofran), oxaliplatin (Eloxatin), palonosetron (Aloxi), pemetrexed (Alimta), pentostatin (Nipent), potassium chloride (KCl), prochlorperazine (Compazine), propofol (Diprivan), ranitidine (Zantac), sodium bicarbonate, thiotepa, topotecan (Hycamtin), vancomycin, vinblastine, vincristine, zidovudine (AZT, Retrovir).
Abraxane
Manufacturer states, “Use of specialized DEHP-free solution containers or administration sets is not necessary.” The use of medical devices containing silicone oil as a lubricant (i.e., syringes and IV bags) to reconstitute and administer Abraxane may result in the formation of proteinaceous strands. Additional specific information not available.
Rate of administration
Conventional paclitaxel
A single dose properly diluted must be equally distributed over 3 hours or as indicated in Usual Dose. Use of an in-line filter not greater than 0.22 microns required. Use a metriset (60 gtt/mL) or an infusion pump appropriate to control flow. Rate extended to 24 hours in some regimens.
Abraxane
A single dose properly reconstituted and equally distributed over 30 minutes.
Actions
All formulations
An antineoplastic. A novel antimicrotubule inhibitor. Paclitaxel derived from the bark of Pacific yew has now been replaced by paclitaxel produced semi-synthetically from a renewable source (needles and twigs of the Himalayan yew). Both are chemically identical. Through specific processes it stabilizes microtubules by preventing depolymerization. This action inhibits the normal dynamic reorganization of the microtubule network essential for vital interphase and mitotic cellular functions. Also induces abnormal bundles of microtubules throughout the cell cycle and multiple asters of microtubules during mitosis. Distribution and/or tissue binding is extensive. Evidence suggests metabolism in the liver via the cytochrome P450 isoenzyme system (CYP2C8 and CYP3A4). Terminal half-life ranges from 13.1 to 27 hours. Excreted primarily as metabolites in feces and, to a lesser extent, in urine.
Conventional paclitaxel
More active in patients who have not received previous chemotherapy.
Abraxane
Consists of albumin-bound paclitaxel nanoparticles. Highly bound to serum proteins; metabolized in the liver, primarily by CYP2C8 and, to a lesser extent, by CYP3A4. Terminal half-life ranges from 13 to 27 hours. Minimal excretion in urine and feces.
Indications and uses
Conventional paclitaxel
First-line and subsequent therapy for the treatment of advanced carcinoma of the ovary. ■ First-line treatment for ovarian cancer in combination with cisplatin. ■ Adjuvant treatment of node-positive breast cancer administered sequentially to standard doxorubicin-containing combination chemotherapy. Most effective in estrogen- and progesterone-receptor–negative tumors. ■ Metastatic breast cancer refractory to initial chemotherapy or for a relapse within 6 months. ■ First-line treatment, in combination with cisplatin, for non–small-cell lung cancer in patients who are not candidates for potentially curative surgery or radiation therapy. ■ Second-line treatment of AIDS-related Kaposi’s sarcoma.
Abraxane
Treatment of breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy. Prior therapy should have included an anthracycline unless clinically contraindicated. ■ First-line treatment of locally advanced or metastatic non–small-cell lung cancer (NSCLC) (in combination with carboplatin) in patients who are not candidates for curative surgery or radiation therapy. ■ First-line treatment of patients with metastatic adenocarcinoma of the pancreas, in combination with gemcitabine.
Unlabeled uses:
Conventional paclitaxel:
Advanced head and neck cancer. ■ Cancers of the bladder and cervix. ■ Small-cell lung cancer. ■ In combination with other agents for treatment of metastatic breast cancer. ■ Relapsed or refractory testicular cancer and testicular germ cell tumors. ■ Treatment of (unknown primary) adenocarcinoma.
Abraxane:
Recurrent ovarian, fallopian, and primary peritoneal cancers.
Contraindications
Conventional paclitaxel
Baseline neutropenia less than 1,500 cells/mm3 in patients with solid tumors or baseline neutropenia less than 1,000 cells/mm3 in patients with AIDS-related Kaposi’s sarcoma. History of prior severe hypersensitivity reactions to paclitaxel or other drugs formulated in polyoxyethylated castor oil (Cremophor EL [e.g., cyclosporine, teniposide]).
Abraxane
Baseline neutrophil count less than 1,500 cells/mm3. ■ Patients who experience a severe hypersensitivity reaction to Abraxane should not be rechallenged with the drug.
Precautions
All formulations
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Usually administered by or under the direction of the physician specialist in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency.
Conventional paclitaxel
Use caution in patients with cardiac conduction abnormalities, CHF, and MI within previous 6 months. ■ Bradycardia, hypertension, and hypotension have been observed but rarely required treatment. Occasionally the infusion must be interrupted or discontinued because of initial or recurrent hypertension; see Monitor. ■ Pre-existing neuropathies resulting from prior therapies are not a contraindication for paclitaxel therapy. ■ Various studies show that incidence and severity of neurotoxicity and hematologic toxicity increase with dose, especially above 190 mg/M2. ■ Use with caution in patients with a total bilirubin greater than 2 times the ULN. May be at increased risk of toxicity, especially profound myelosuppression; see Dose Adjustments. ■ See Drug/Lab Interactions.
Abraxane
Do not substitute for or with other paclitaxel formulations. ■ Use of gloves recommended. Wash skin immediately if contact occurs. Flush mucous membranes thoroughly with water if contact occurs. Topical exposure may result in tingling, burning, and redness. ■ Premedication to prevent hypersensitivity reactions is not required but may be needed in patients who have had a prior hypersensitivity reaction to Abraxane. Reports of severe and sometimes fatal hypersensitivity reactions have occurred; see Contraindications; do not rechallenge patients who have had a severe reaction. ■ Neutropenia is dose dependent and is a dose-limiting toxicity. Do not administer Abraxane to patients with a baseline absolute neutrophil count (ANC) of less than 1,500 cells/mm3; see Dose Adjustments. ■ Sensory neuropathy is dose dependent and schedule dependent and occurs frequently; see Dose Adjustments. ■ Sepsis occurred in 5% of patients, with or without neutropenia, who received Abraxane in combination with gemcitabine. Biliary obstruction or the presence of a biliary stent were risk factors for severe or fatal sepsis. ■ Pneumonitis, including some cases that were fatal, has been reported in patients receiving Abraxane in combination with gemcitabine; see Antidote. ■ Has not been studied in patients with severe renal dysfunction or end-stage renal disease. ■ Use with caution in patients with hepatic impairment. May be at increased risk for toxicity, particularly myelosuppression; see Dose Adjustments. ■ Derived from human albumin; may carry a risk of transmission of viral disease or Creutzfeldt-Jakob disease; risk considered extremely remote. ■ Has not been studied in patients who have had a hypersensitivity reaction to conventional paclitaxel.
Monitor: All formulations:
Neutropenia is dose dependent and is the dose-limiting toxicity. Obtain baseline CBC with differential and platelet count. Monitor frequently during therapy and before each dose. ■ Monitor injection site carefully; avoid extravasation. ■ Observe closely for signs of infection. Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. ■ Use prophylactic antiemetics to reduce nausea and vomiting and increase patient comfort. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood).
Conventional paclitaxel:
Neutrophil nadir occurs around Day 11; see Dose Adjustments. ■ Monitor VS frequently, particularly during the first hour of the infusion. ■ Consider obtaining a baseline ECG; arrhythmias have occurred. Continuous cardiac monitoring required for all patients with an abnormal baseline ECG or for those who experienced conduction arrhythmias during administration of a previous dose. ■ Monitoring of cardiac function is recommended when paclitaxel is used in combination with doxorubicin; see doxorubicin monograph. ■ Anaphylaxis and severe hypersensitivity reactions characterized by dyspnea, hypotension requiring treatment, angioedema, and generalized urticaria have been reported. Fatal reactions have occurred despite premedication. ■ Most severe hypersensitivity reactions occur in the first hour; chest pain, dyspnea, flushing, and tachycardia were the most frequent initial symptoms; abdominal pain, diaphoresis, extremity pain, and hypertension also occurred. Monitor all vital signs, including BP, continuously for the first 30 minutes of the infusion and at frequent intervals after that. Incidence seems to decrease with subsequent doses. ■ Treatment can often be continued in patients with mild hypersensitivity reactions if proper premedication is given. ■ Monitor injection site carefully; avoid extravasation. Incidence of inflammation increased with 24-hour infusions. Injection site reactions may occur during administration or be delayed by 7 to 10 days. Recurrence of skin reactions at a site of previous extravasation following administration of paclitaxel (“recall”) has been reported.
Abraxane:
Limited infusion time (30 minutes) reduces the likelihood of infusion-related reactions; however, they have been reported; monitor for S/S. ■ Monitor for S/S of hypersensitivity reactions. ■ Monitor for S/S of sensory neuropathy. ■ Monitor for S/S of pneumonitis. ■ Based on patient history, a baseline ECG may be indicated. ■ See Precautions and Dose Adjustments.
Patient education: All formulations:
Males and females should avoid conception; nonhormonal birth control recommended. ■ Review of monitoring requirements and adverse events before therapy imperative. ■ Report any unusual or unexpected symptoms, side effects, pain or burning at injection site, S/S of a hypersensitivity reaction (e.g., bronchospasm, difficulty breathing, rash, urticaria), signs of infection (e.g., chills, fever, night sweats), signs of sensory neuropathy (e.g., numbness, tingling, or burning in hands and/or feet), signs of pneumonitis (e.g., dry, persistent cough or shortness of breath), or signs of bleeding (e.g., bruising, tarry stools, blood in urine, pinpoint red spots on skin) as soon as possible. ■ Avoid tasks that require mental alertness (e.g., driving, operating machinery) until the effect of the medication is known. Side effects such as fatigue, lethargy, and malaise may affect the ability to perform these tasks. ■ See Appendix D, p. 1333. ■ Obtain name and telephone number of a contact person for emergencies, questions, or problems. ■ Seek resources for counseling or supportive therapy. ■ Manufacturer provides a patient information booklet.
Maternal/child: All formulations:
Category D: females should avoid pregnancy, and males should avoid fathering a child. May cause fetal harm. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Conventional paclitaxel:
CNS toxicity (rarely associated with death) was reported in one pediatric trial using high-dose paclitaxel. Use of antihistamines and the ethanol contained in the paclitaxel may have contributed to toxicity noted.
Elderly: All formulations:
Studies suggest response is similar to that seen in younger patients.
Conventional paclitaxel:
Incidence of side effects, including myelosuppression, neuropathy, and cardiovascular events, may be increased in the elderly.
Abraxane:
Increased incidence of dehydration, diarrhea, epistaxis, fatigue, and peripheral edema was seen in elderly patients receiving Abraxane as monotherapy for MBC. Incidence of myelosuppression, neuropathy, and arthralgia was more common in patients receiving Abraxane and carboplatin for treatment of NSCLC. Incidence of decreased appetite, dehydration, diarrhea, and epistaxis was more common in patients receiving Abraxane and gemcitabine for treatment of adenocarcinoma of the pancreas.
Drug/lab interactions
All formulations
Do not administer chloroquine or live virus vaccines to patients receiving antineoplastic agents.
Conventional paclitaxel
Formal drug interaction studies have not been conducted. To reduce potential for profound myelosuppression when using paclitaxel and cisplatin concurrently, give paclitaxel first, then cisplatin. ■ Neurotoxicity and symptomatic motor dysfunction occurring with higher doses (greater than 250 mg/M2) may be potentiated by cisplatin and filgrastim (G-CSF). ■ May cause additive effects with bone marrow–suppressing agents, radiation therapy, or agents that cause blood dyscrasias (e.g., amphotericin B, antithyroid agents [methimazole (Tapazole)], azathioprine, chloramphenicol, ganciclovir [Cytovene], interferon, plicamycin [Mithracin], zidovudine [AZT, Retrovir]). Reduced doses may be required. ■ May increase levels of doxorubicin and its active metabolite when drugs are used in combination. ■ Metabolized by cytochrome P450 isoenzymes CYP3A4 and CYP2C8. Use caution when administered concomitantly with known substrates (e.g., buspirone [BuSpar], eletriptan [Relpax], felodipine [Plendil], lovastatin [Mevacor], midazolam [Versed], sildenafil [Viagra, Revatio], simvastatin [Zocor], and triazolam [Halcion]), inhibitors (e.g., atazanavir [Reyataz], clarithromycin [Biaxin], indinavir [Crixivan], itraconazole [Sporanox], ketoconazole [Nizoral], nefazodone, nelfinavir [Viracept], ritonavir [Norvir], saquinavir [Invirase], and telithromycin [Ketek]), and inducers (e.g., rifampin [Rifadin] and carbamazepine [Tegretol]) of CYP3A4. ■ Other medications that are substrates and/or inducers of CYP3A4 (e.g., ritonavir [Norvir], saquinavir [Invirase], indinavir [Crixivan], and nelfinavir [Viracept]) may alter the metabolism of paclitaxel but have not been evaluated in clinical trials. ■ Use caution when administered concomitantly with known substrates (e.g., repaglinide [Prandin], rosiglitazone [Avandia]), inhibitors (e.g., gemfibrozil [Lopid]), and inducers (e.g., rifampin [Rifadin]) of CYP2C8. ■ Dexamethasone (Decadron), diphenhydramine (Benadryl), cimetidine (Tagamet), and ranitidine (Zantac) do not affect the protein binding of paclitaxel.
Abraxane
Drug interaction studies have not been conducted. See All Formulations above. ■ Use caution when administering with medicines known to inhibit or induce CYP2C8 or CYP3A4. Drugs that may inhibit these enzymes include ketoconazole (Nizoral) and other imidazole antifungals, erythromycin, fluoxetine (Prozac), gemfibrozil (Lopid), cimetidine (Tagamet), ritonavir (Norvir), saquinavir (Invirase), indinavir (Crixivan), and nelfinavir (Viracept). Drugs that may induce these enzymes include rifampin (Rifadin), carbamazepine (Tegretol), phenobarbital, phenytoin (Dilantin), efavirenz (Sustiva), nevirapine (Viramune).
Side effects
Conventional paclitaxel
Dose dependent and generally reversible, but may be fatal. All patients were premedicated to prevent hypersensitivity reactions. Abnormal ECG, alopecia, arthralgia/myalgia, asthenia, autonomic neuropathy resulting in paralytic ileus, bleeding, bone marrow suppression (anemia, leukopenia, neutropenia, thrombocytopenia), bradycardia, CHF (including cardiac dysfunction and reduction in left ventricular ejection fraction or ventricular failure [more common in patients receiving combination therapy with anthracyclines]), diarrhea, elevated alkaline phosphatase, elevated AST, elevated bilirubin, febrile neutropenia, fever, fluid retention and edema, hypersensitivity reactions (moderate [e.g., dyspnea, flushing, hypotension, rash, tachycardia]; severe [e.g., chest pain, dyspnea requiring bronchodilators, hypotension requiring treatment, generalized urticaria]), hypertension, hypotension, infections including opportunistic infections (chills, fever, night sweats), injection site reactions (cellulitis, fibrosis, induration, necrosis, phlebitis, skin exfoliation), mucositis, nausea and vomiting, numbness, optic nerve and/or visual disturbances, peripheral neuropathy, respiratory reactions (interstitial pneumonia, lung fibrosis, pleural effusions, pulmonary embolism, respiratory failure), Stevens-Johnson syndrome, toxic epidermal necrolysis, and visual disturbances. A grand mal seizure occurred in one patient. Other side effects (e.g., cardiac arrest, cardiac ischemia/infarction, CVA, hepatic necrosis, and hepatic encephalopathy leading to death; intestinal obstruction; intestinal perforation; ischemic colitis; pancreatitis; thrombosis/embolism) have been reported rarely. A higher incidence of elevated liver function tests and renal toxicity is seen in Kaposi’s sarcoma patients.
Post-marketing:
Diffuse edema, thickening, and sclerosing of the skin; exacerbation of S/S of scleroderma; ototoxicity.
Abraxane
Single-agent use in patients with MBC:
Abnormal ECG, alopecia, anemia, diarrhea, elevation of alkaline phosphate and AST, fatigue/asthenia, infections (oral candidiasis, pneumonia, and respiratory tract), myalgia/arthralgia, nausea, neutropenia, and sensory neuropathy are most common.
Combination use in patients with NSCLC:
Alopecia, anemia, fatigue, nausea, neutropenia, peripheral neuropathy, and thrombocytopenia are most common. The most serious side effects reported are anemia and pneumonia. The most common side effects that result in dose reduction, the withholding of a dose, or a delay in dosing are anemia, neutropenia, and thrombocytopenia. Neutropenia, peripheral neuropathy, and thrombocytopenia sometimes resulted in permanent discontinuation of Abraxane.
Combination use in patients with adenocarcinoma of the pancreas:
Alopecia, decreased appetite, dehydration, diarrhea, fatigue, fever, nausea, neutropenia, peripheral edema, peripheral neuropathy, rash, and vomiting. The most common serious side effects are dehydration, fever, pneumonia, and vomiting. The most common side effects that result in dose reduction or delay in dosing are anemia, diarrhea, fatigue, neutropenia, peripheral neuropathy, and thrombocytopenia. The most common side effects resulting in permanent discontinuation are fatigue, peripheral neuropathy, and thrombocytopenia.
All diagnoses with abraxane:
Hypersensitivity reactions have occurred but are not usually severe; premedication is not indicated. Other side effects have been reported and include bradycardia, cardiovascular events (e.g., cardiac ischemia/infarction, SVT), dehydration, extravasation, fever, hypotension, pancytopenia, pneumonitis, pneumothorax, Stevens-Johnson syndrome, toxic epidermal necrolysis, and many others. Frequency of sensory neuropathy may increase with cumulative dose.
Post-marketing:
Many of these side effects occur with paclitaxel and Abraxane and include CHF, left ventricular dysfunction, and atrioventricular block; cranial nerve palsies; hepatic necrosis and hepatic encephalopathy; injection site reactions; interstitial pneumonia; intestinal obstruction or perforation; ischemic colitis; pancreatitis; paralytic ileus; pneumonitis; pulmonary embolism; visual disorders (reduced visual acuity); vocal cord paresis. Rare reports of severe hypersensitivity reactions have occurred with Abraxane.
Antidote
Keep physician informed of all side effects. Most will be treated symptomatically as indicated. Most hypersensitivity reactions will subside with temporary discontinuation of paclitaxel, and incidence seems to decrease with subsequent doses. Moderate reactions such as dyspnea, flushing, hypotension, skin reactions, or tachycardia do not usually require interruption of treatment. Severe reactions may require epinephrine (Adrenalin), antihistamines (e.g., diphenhydramine [Benadryl]), corticosteroids (e.g., dexamethasone [Decadron]), or bronchodilators (e.g., albuterol, theophylline [aminophylline]). Most severe reactions should not be rechallenged, but some patients tolerated subsequent doses of conventional paclitaxel; see Contraindications. Neutropenia can be profound, and the nadir usually occurs about Day 11 with conventional paclitaxel. Recovery is generally rapid and spontaneous but may be treated with filgrastim (Neupogen, Zarxio), pegfilgrastim (Neulasta). Severe thrombocytopenia (nadir Day 8 or 9 with conventional paclitaxel) may require platelet transfusions. Severe anemia (less than 8 Gm/dL) may require packed cell transfusions. Hypotension and bradycardia do not usually occur at the same time except in hypersensitivity. Treat only if symptomatic. Some arrhythmias (e.g., nonspecific repolarization abnormalities, sinus tachycardia, and PVCs) are common and may not require intervention. Promptly treat any serious or symptomatic arrhythmia (e.g., conduction abnormalities, ventricular tachycardia), and monitor continuously during subsequent doses. Neurologic symptoms tend to worsen with each course; see Dose Adjustments. Usually improve within several months. Severe peripheral neuropathies or seizure may necessitate discontinuation of paclitaxel. Permanently discontinue treatment with Abraxane and gemcitabine if pneumonitis develops. There is no specific antidote for overdose. Supportive therapy will help sustain the patient in toxicity. Resuscitate if indicated.
Palifermin
(PAL-lih-fur-min)
Kepivance, KGF
Growth factor
pH 6.5
Usual dose
Administered as an IV bolus injection for 3 consecutive days before and 3 consecutive days after myelotoxic therapy (a total of 6 doses). Do not administer within 24 hours before, during infusion of, or within 24 hours after administration of myelotoxic chemotherapy. Has resulted in increased severity and duration of oral mucositis. Myelotoxic therapy is high-dose chemotherapy, with or without radiation, that is destructive to bone marrow or any of its components. Followed by bone marrow transplant/hematopoietic stem cell support.
Pre-myelotoxic therapy (first 3 doses):
Administer 60 mcg/kg/day for 3 consecutive days before beginning myelotoxic therapy, with the third dose 24 to 48 hours before myelotoxic therapy.
Post-myelotoxic therapy (last 3 doses):
Administer 60 mcg/kg/day for 3 consecutive days after myelotoxic therapy is complete; the first of these doses should be administered after, but on the same day of, hematopoietic stem cell infusion and at least 4 days after the most recent administration of palifermin. See Precautions.
Dose adjustments
None indicated. Gender-related differences were not observed. ■ No dose adjustment is recommended in impaired renal function. ■ Pharmacokinetic studies have not been performed for pediatric patients or for patients with hepatic insufficiency.
Dilution
Available as a lyophilized powder in a 6.25-mg single vial. Reconstitute by slowly injecting 1.2 mL SWFI into vial. Final concentration is 5 mg/mL. Swirl contents gently. Do not shake. Dissolution should take less than 3 minutes. Solution is clear and colorless. Do not use if particulates are present or if solution is discolored.
Filters:
Manufacturer states, “Do not filter the reconstituted solution during preparation or administration.”
Storage:
Keep vials in carton until use. Store at 2° to 8° C (36° to 46° F). Protect from light. Do not use beyond expiration date on vial. Reconstituted product should be used immediately but can be stored up to 24 hours if refrigerated and stored in its carton. Do not freeze. Solution may be warmed to RT before injection but should not be left at RT for more than 1 hour and must be protected from light.
Compatibility
Specific information not available; however, manufacturer states, “If heparin is used to maintain an IV line, saline should be used to flush the line before and after administration since palifermin has been shown to bind to heparin in vitro.”
Rate of administration
A single dose administered as an IV bolus injection. If heparin is used to maintain an IV line, flush the line with NS before and after administration.
Actions
Human keratinocyte growth factor (KGF) produced by recombinant DNA technology. Binding of KGF to its receptor results in proliferation, differentiation, and migration of epithelial cells. KGF receptors are present on the epithelial cells in many tissues, including the tongue, buccal mucosa, esophagus, stomach, intestine, salivary gland, lung, liver, pancreas, kidney, bladder, mammary gland, skin (hair follicles and sebaceous gland), and the lens of the eye. The KGF receptor is not present on the cells of the hematopoietic lineage. KGF stimulates the growth of cells in tissues such as the skin and the epithelial layer of the mouth, stomach, and colon. Protects the epithelial cells that line the mouth and GI tract from the damage caused by chemotherapy and radiation and stimulates the growth and development of new epithelial cells. Average half-life is 4.5 hours (range: 3.3 to 5.7 hours).
Indications and uses
To decrease the incidence and duration of severe oral mucositis in patients with hematologic malignancies who are receiving myelotoxic therapy (high-dose chemotherapy), with or without radiation therapy, and who require hematopoietic stem cell support. Indicated as supportive care for preparative regimens predicted to result in equal to or greater than WHO Grade 3 mucositis in the majority of patients.
Limitations of use:
Safety and efficacy for use in patients with nonhematologic malignancies not established; see Precautions. ■ Not recommended for use with melphalan 200 mg/M2 as a conditioning regimen.
Investigational uses:
Studies to determine whether palifermin can be used safely in other types of cancer are in progress.
Contraindications
Manufacturer states, “None.”
Precautions
Safety and efficacy have not been established in patients with nonhematologic malignancies. Effect of palifermin on stimulation of KGF receptor–expressing, nonhematopoietic tumors in patients is not known. There is some evidence of tumor growth and stimulation in cell cultures and in animal models of nonhematopoietic human tumors.
Monitor:
Monitor improvement in symptoms of oral mucositis. ■ Monitor for the appearance of mucocutaneous adverse effects (e.g., edema, erythema, oral/perioral dysesthesia [impairment of sensitivity to touch], rash, taste alteration, tongue discoloration, tongue thickening).
Patient education:
Review possible side effects. ■ Promptly report side effects (e.g., edema, impairment of sensitivity to touch [especially around the mouth], rash, taste alteration, tongue discoloration, tongue thickening). ■ Inform patient of the evidence of tumor growth and stimulation in cell cultures and animal models of nonhematopoietic human tumors.
Maternal/child:
Category C: potential benefit to mother must justify potential risk to fetus. ■ Discontinue breast-feeding. ■ Information on dosing and safety in pediatric patients is limited. However, use in pediatric patients ages 1 to 16 years is supported by evidence from well-controlled studies in adults and from a Phase 1 study that included 27 pediatric patients with acute leukemia undergoing hematopoietic stem cell transplant. Adverse events were similar to those reported for adults, and age did not affect the pharmacokinetics of palifermin.
Elderly:
Age-related differences have not been observed.
Drug/lab interactions
Formal studies have not been conducted. ■ Interacts with unfractionated as well as low-molecular-weight heparins (e.g., enoxaparin [Lovenox]). When coadministered, there was no significant effect of palifermin on heparin activity with respect to aPPT. However, coadministration resulted in an increased palifermin AUC and a decreased palifermin clearance, volume of distribution, and half-life. If heparin is used to maintain an IV line, NS should be used to flush the line before and after palifermin administration. ■ Do not administer within 24 hours before, during infusion of, or within 24 hours after administration of myelotoxic chemotherapy. Has resulted in increased severity and duration of oral mucositis.
Side effects
The most common serious side effects are fever, GI events, respiratory events, and skin rash. Other commonly reported reactions include arthralgia, edema, elevated serum amylase and lipase, hypertension, oral toxicities (alteration of taste, oral/perioral dysesthesia, tongue discoloration, tongue thickening), pain, paresthesia, proteinuria, and skin toxicities (erythema, pruritus, rash). Has the potential for immunogenicity, but the clinical significance is unknown.
Post-marketing:
Anaphylaxis, cataracts, edema of the face and mouth, palmar-plantar erythrodysesthesia syndrome (dysesthesia, edema on the palms and soles, erythema), tongue disorders (e.g., bumps, edema, redness), transient hyperpigmentation of the skin, vaginal edema and erythema.
Antidote
Notify physician of all side effects. Most will be treated symptomatically.
Palonosetron
(pal-oh-NOH-seh-tron)
Aloxi
Antiemetic (5-HT3 receptor antagonist)
pH 4.5 to 5.5
Usual dose
Chemotherapy-induced nausea and vomiting:
A single dose of 0.25 mg approximately 30 minutes before the start of chemotherapy. Has been coadministered with corticosteroids (e.g., dexamethasone [Decadron]) and metoclopramide (Reglan); see Drug/Lab Interactions.
Postoperative nausea and vomiting:
A single dose of 0.075 mg immediately before induction of anesthesia.
Pediatric dose
Chemotherapy-induced nausea and vomiting:
A single dose of 20 mcg/kg (maximum 1.5 mg) approximately 30 minutes before the start of chemotherapy.
Dose adjustments
No dose adjustment required based on age or race or in patients with any degree of renal or hepatic impairment.
Dilution
Available in 0.25 mg/5 mL and 0.075 mg/1.5 mL single-use vials. May be given undiluted.
Filters:
No data available from manufacturer.
Storage:
Before use, store at CRT. Protect from freezing and light.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Should not be mixed with other drugs. Flush the infusion line with NS before and after administration.”
One source suggests the following compatibilities:
Additive:
Not recommended by manufacturer.
Dexamethasone (Decadron).
Y-site:
Ampicillin/sulbactam (Unasyn), atropine, carboplatin (Paraplatin), cefazolin (Ancef), cefotetan, cisatracurium (Nimbex), cisplatin, cyclophosphamide (Cytoxan), dacarbazine (DTIC), docetaxel (Taxotere), famotidine (Pepcid IV), fentanyl, fluorouracil (5-FU), gemcitabine (Gemzar), gentamicin, glycopyrrolate (Robinul), heparin, hetastarch in electrolytes (Hextend), hydromorphone (Dilaudid), ifosfamide (Ifex), irinotecan (Camptosar), lidocaine, lorazepam (Ativan), mannitol (Osmitrol), meperidine (Demerol), metoclopramide (Reglan), metronidazole (Flagyl IV), midazolam (Versed), morphine, neostigmine, oxaliplatin (Eloxatin), paclitaxel (Taxol), potassium chloride, promethazine (Phenergan), rocuronium (Zemuron), succinylcholine (Anectine), sufentanil (Sufenta), topotecan (Hycamtin), vancomycin, vecuronium.
Rate of administration
Flush the infusion line with NS before and after administration.
Chemotherapy-induced nausea and vomiting in adults:
A single dose as an IV injection equally distributed over 30 seconds.
Chemotherapy-induced nausea and vomiting in pediatric patients:
A single dose as an IV infusion equally distributed over 15 minutes.
Postoperative nausea and vomiting:
A single dose as an IV injection equally distributed over 10 seconds.
Actions
A long-acting (up to 120 hours [5 days]) antinauseant and antiemetic agent. A selective antagonist of specific serotonin (5HT3) receptors. Has a strong binding affinity for this receptor and little or no affinity for other receptors. Chemotherapeutic agents such as cisplatin increase the release of serotonin from specific cells in the GI tract, causing emesis. Postoperative nausea and vomiting is also triggered by the release of serotonin. By antagonizing serotonin receptors both on the vagus nerve in the periphery and centrally in the chemoreceptor trigger zone, the incidence and severity of chemotherapy-induced nausea and vomiting and postoperative nausea and vomiting are decreased. 62% bound to plasma protein. Partially metabolized by multiple CYP enzymes; however, palonosetron is not an inhibitor or an inducer of CYP enzyme activity. Eliminated slowly from the body through both metabolic pathways and renal excretion. Has a prolonged half-life of approximately 40 hours. 80% of a single dose was recovered in urine within 144 hours.
Indications and uses
Prevention of acute nausea and vomiting associated with initial and repeat courses of moderately and highly emetogenic cancer chemotherapy in pediatric patients 1 month to less than 17 years of age and in adults. ■ Prevention of delayed nausea and vomiting associated with initial and repeat courses of moderately emetogenic cancer chemotherapy in adults. Studies identified cisplatin in doses equal to or greater than 70 mg/M2 and cyclophosphamide (Cytoxan) in doses equal to or greater than 1,100 mg/M2 as highly emetogenic. Studies identified cisplatin in doses equal to or less than 50 mg/M2, cyclophosphamide in doses less than 1,100 mg/M2, doxorubicin (Adriamycin) in doses greater than 25 mg/M2, methotrexate in doses greater than 250 mg/M2, and carboplatin (Paraplatin), epirubicin (Ellence), and irinotecan (Camptosar) in standard doses as moderately emetogenic. ■ Prevention of postoperative nausea and vomiting (PONV) in adults for up to 24 hours following surgery. Efficacy beyond 24 hours has not been demonstrated.
Contraindications
Known hypersensitivity to palonosetron or any of its components. ■ See Precautions.
Precautions
Hypersensitivity reactions, including anaphylaxis, have been reported with or without known hypersensitivity to other 5-HT3 receptor antagonists. Cross-sensitivity may occur with other selective 5-HT3 receptor antagonists (e.g., dolasetron [Anzemet], granisetron [Kytril], ondansetron [Zofran]). ■ Serotonin syndrome has been reported with 5-HT3 receptor antagonists. Most reports were associated with concomitant use of serotonergic drugs (e.g., selective serotonin reuptake inhibitors [e.g., paroxetine (Paxil), escitalopram (Lexapro)], serotonin and norepinephrine reuptake inhibitors [e.g., duloxetine (Cymbalta), venlafaxine (Effexor XR)], monoamine oxidase inhibitors [e.g., selegiline (Eldepryl)], mirtazapine [Remeron], fentanyl, lithium, tramadol [Ultram], and intravenous methylene blue). Some cases were fatal. ■ Routine prophylaxis is not recommended for patients in whom there is little expectation of PONV. However, for patients in whom nausea and vomiting must be avoided during the postoperative period, prophylaxis is recommended even when the incidence of PONV is low. ■ Palonosetron does not appear to have any effect on ECG intervals, including QTC duration.
Monitor:
Observe closely. Monitor VS. ■ Monitor for serotonin syndrome, especially when palonosetron is used concurrently with other serotonergic drugs. Symptoms associated with serotonin syndrome may include the following combination of S/S: mental status changes (e.g., agitation, coma, delirium, hallucinations), autonomic instability (e.g., diaphoresis, dizziness, flushing, hyperthermia, labile blood pressure, tachycardia), neuromuscular symptoms (e.g., hyperreflexia, incoordination, myoclonus, rigidity, tremor), and seizures, with or without GI symptoms (e.g., diarrhea, nausea, vomiting). ■ Ambulate slowly to avoid orthostatic hypotension. ■ Stool softeners or laxatives may be required to prevent constipation.
Patient education:
Request assistance with ambulation. ■ Used to prevent and/or treat both early and late N/V caused by chemotherapy. Report persistent N/V promptly. ■ Maintain adequate hydration. ■ Review prescription and nonprescription medications with health care provider. Drug interactions are possible, especially with diuretics or antiarrhythmics. ■ Review other medical conditions with health care provider. ■ Promptly report S/S of serotonin syndrome (e.g., altered mental status, autonomic instability, and neuromuscular symptoms); see Precautions.
Maternal/child:
Category B: use during pregnancy only if clearly needed. ■ Safety for use during breast-feeding not established; effects unknown, but potential for tumorigenicity is a concern. Discontinue breast-feeding. ■ Safety and effectiveness have been established for use in pediatric patients 1 month to less than 17 years of age for the prevention of acute nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy, including highly emetogenic chemotherapy. Pediatric patients require a higher palonosetron dose than adults to prevent chemotherapy-induced nausea and vomiting. However, the safety profile seen in pediatric patients is consistent with the established profile in adults. ■ Safety and effectiveness have not been established in pediatric patients for prevention of postoperative nausea and vomiting.
Elderly:
Safety and effectiveness similar to younger adults; however, greater sensitivity in some elderly cannot be ruled out. No dose adjustment or special monitoring required.
Drug/lab interactions
Eliminated through both renal excretion and metabolic pathways. Does not induce or inhibit the cytochrome P450 drug metabolizing system; potential for clinically significant drug interactions is considered to be low. ■ Serotonin syndrome (including altered mental status, autonomic instability, and neuromuscular symptoms) has been reported following the concomitant use of 5-HT3 receptor antagonists and other serotonergic drugs, including selective serotonin reuptake inhibitors (SSRIs) and serotonin and norepinephrine reuptake inhibitors (SNRIs); see Precautions. ■ Has been safely administered with analgesics, antiemetics/antinauseants, antispasmodics, anticholinergic agents, and corticosteroids. ■ Has been safely administered with PO metoclopramide (10 mg 4 times daily). ■ Has been administered with oral aprepitant (Emend). ■ Does not appear to inhibit the antitumor activity of emetogenic cancer chemotherapies (cisplatin, cyclophosphamide, cytarabine, doxorubicin, and mitomycin C have been tested in murine tumor models). ■ See Precautions.
Side effects
Chemotherapy-induced nausea and vomiting in adults:
The most common side effects are headache and constipation. Other reported side effects include abdominal pain, diarrhea, dizziness, fatigue, and insomnia.
Chemotherapy-induced nausea and vomiting in pediatric patients:
Allergic dermatitis, dizziness, dyskinesia, headache, and infusion site pain.
Postoperative nausea and vomiting:
The most common side effects, occurring in at least 2% of patients, are bradycardia, constipation, headache, and QT prolongation. Several other side effects are reported in 1% or fewer of patients.
Overdose:
Doses more than 20 times the recommended dose did not cause significant problems. Collapse, convulsions, cyanosis, gasping, and pallor occurred in animal studies with rats and mice.
Post-marketing:
Rare cases of hypersensitivity reactions, including anaphylaxis, anaphylactic shock, and injection site reactions (burning, induration, pain). Serotonin syndrome has been reported as a 5-HT3 class reaction.
Antidote
Most side effects will be treated symptomatically. Keep physician informed. There is no specific antidote. If symptoms of serotonin syndrome occur, discontinue palonosetron and initiate supportive care. Has a large volume of distribution; dialysis not likely to be effective in overdose. Treat hypersensitivity reactions and resuscitate as indicated.
Pamidronate disodium
(pah-MIH-droh-nayt DYE-so-dee-um)
APD, Aredia
Bone resorption inhibitor
Antihypercalcemic (bisphosphonate)
pH 6 to 7.4
Usual dose
Prehydration required. Do not exceed a 90-mg dose. See Precautions and Monitor.
Moderate hypercalcemia (corrected serum calcium of 12 to 13.5 mg/dL):
One dose of 60 to 90 mg as an infusion.
Severe hypercalcemia (corrected serum calcium greater than 13.5 mg/dL):
One dose of 90 mg as an infusion. Serum calcium levels should fall into the normal range (8.5 to 10.5 mg/100 mL [1 dL], corrected for serum albumin).
Experience is limited, but retreatment with the same dose may be considered if hypercalcemia is not fully corrected or recurs; wait at least 7 days from completion of first infusion to allow full response. Always used in conjunction with adequate hydration and appropriate testing. See Precautions/Monitor.
Paget’s disease:
30 mg/day as an infusion for 3 consecutive days (total dose is 90 mg over 3 days). Selected patients have been retreated with the same dose when indicated. Experience limited. Prehydration required; see Precautions and Monitor.
Osteolytic bone lesions of multiple myeloma:
90 mg as an infusion once every 30 days. Optimal duration of therapy not known. Withhold dose if renal function has deteriorated; see Dose Adjustments, Precautions, and Monitor.
Osteolytic bone metastases of breast cancer:
90 mg as an infusion every 3 to 4 weeks. Optimum duration of therapy not known. Withhold dose if renal function has deteriorated; see Dose Adjustments, Precautions, and Monitor.
Dose adjustments
Accumulation of pamidronate in renally impaired patients is not anticipated if dosed on a monthly schedule. No experience with creatinine above 5 mg/100 mL (1 dL) or in severe hepatic disease. ■ See Precautions and Elderly.
Osteolytic bone lesions of multiple myeloma and osteolytic bone metastases of breast cancer:
Withhold dose if renal function has deteriorated. Renal deterioration is defined as an increase of 0.5 mg/dL in patients with a normal baseline creatinine or an increase of 1 mg/dL in patients with abnormal baseline creatinine. One study suggests that treatment should not be resumed until SCr has returned to within 10% of baseline value.
Dilution
Reconstitute each 30- or 90-mg vial with 10 mL SWFI. Dissolve completely (3 or 9 mg/mL).
Hypercalcemia of malignancy:
Further dilute a single dose in 1,000 mL NS, 1/2NS, or D5W. A minimum of 500 mL diluent may be used if absolutely necessary in patients with compromised cardiovascular status.
Paget’s disease:
Further dilute a single daily dose in 500 mL NS, 1/2NS, or D5W.
Osteolytic bone lesions of multiple myeloma:
Further dilute each 90-mg dose in 500 mL NS, 1/2NS, or D5W.
Osteolytic bone metastases of breast cancer:
Further dilute each 90-mg dose in 250 mL NS, 1/2NS, or D5W.
Storage:
Before reconstitution, store at CRT. After reconstitution, may be refrigerated for up to 24 hours. Stable after dilution for 24 hours at room temperature.
Compatibility
Manufacturer states, “Should be given in a single intravenous solution and line separate from all other drugs, and do not mix with calcium-containing solutions (e.g., Ringer’s solutions).”
Rate of administration
Use of a microdrip (60 gtt/mL) or an infusion pump recommended for even distribution. Do not exceed recommended rate of infusion. Duration should be no less than 2 hours. Too-rapid infusion rate may lead to overdose, elevated BUN and creatinine levels, and renal tubular necrosis. Rate recommendations vary considerably. They are based on specific clinical trials for each diagnosis. In some trials a rate of up to 1 mg/min has been used with caution.
Hypercalcemia of malignancy:
A 60-mg dose or 90-mg dose equally distributed over 2 to 24 hours. Longer infusion times (i.e., greater than 2 hours) may reduce the risk of renal toxicity, particularly in patients with pre-existing renal insufficiency.
Paget’s disease:
A single dose over 4 hours.
Osteolytic bone lesions of multiple myeloma:
A single dose over 4 hours.
Osteolytic bone metastases of breast cancer:
A single dose over 2 hours.
Actions
A bisphosphonate hypocalcemic agent. Reduces serum calcium concentrations by inhibiting bone resorption. Binds to preformed bone surfaces and may block bone mineral dissolution. May also inhibit osteoclast activity. Effectively inhibits accelerated bone resorption resulting from osteoclast hyperactivity induced by various tumors. Does not inhibit bone formation and mineralization. Some reduction in calcium levels seen in 24 to 48 hours, and maximum response in 4 to 7 days. Rapidly adsorbed by bone. Is not metabolized. Half-life is approximately 21 to 35 hours. Slowly excreted in urine.
Indications and uses
Treatment of moderate to severe hypercalcemia of malignancy in patients with or without bone metastasis, in conjunction with adequate hydration. Symptoms of hypercalcemia may include anorexia, bone pain, confusion, constipation, dehydration, depression, fatigue, lethargy, muscle weakness, nausea and vomiting, and polyuria. Severe dehydration may lead to renal insufficiency. With high levels of serum calcium, cardiac manifestations (e.g., bradycardia, cardiac arrest, ventricular arrhythmias) and neurologic symptoms (e.g., coma, seizures, and death) may occur. ■ Treatment of Paget’s disease. ■ Adjunct in treatment of osteolytic lesions of multiple myeloma and osteolytic bone metastases of breast cancer.
Unlabeled uses:
Prevention of bone loss associated with androgen deprivation therapy in prostate cancer.
Contraindications
Hypersensitivity to pamidronate or other bisphosphonates (e.g., alendronate [Fosamax], risedronate [Actonel], zoledronic acid [Reclast, Zometa]).
Precautions
Calcium is bound to albumin. Total serum calcium levels in patients who have hypercalcemia of malignancy may not reflect the severity of the hypocalcemia because concomitant hypoalbuminemia is commonly present. Measurement with ionized calcium levels is preferred. If unavailable, all calcium measurement should be corrected for albumin to establish a basis for treatment and evaluation of treatment. ■ Mild or asymptomatic hypercalcemia will be treated with conservative measures (e.g., saline hydration, with or without diuretics [after correcting hypovolemia]). Consider patient’s cardiovascular status. Corticosteroids may be indicated if the underlying cancer is sensitive (e.g., hematologic cancers). ■ May be used adjunctively with chemotherapy, radiation, or surgery. ■ May cause renal toxicity. Deterioration in renal function progressing to renal failure has been reported and has occurred after the initial or a single dose of pamidronate. Patients with pre-existing renal impairment may be at increased risk for developing toxicity. Do not exceed dose of 90 mg. ■ Osteonecrosis of the jaw (ONJ) has been reported in patients receiving bisphosphonates. The majority of cases have been in cancer patients. Risk factors include cancer, concomitant therapy (e.g., chemotherapy, radiotherapy, corticosteroids), and comorbid conditions (e.g., anemia, coagulopathies, infection, pre-existing oral disease). Literature and case reports suggest a higher frequency of ONJ based on tumor type (breast cancer, multiple myeloma) and dental status (dental extraction, periodontal disease, local trauma, including poorly fitting dentures). Cancer patients should maintain good oral hygiene. Consider dental exam and appropriate preventive dentistry before beginning therapy with bisphosphonates. Avoid invasive dental procedures during bisphosphonate therapy. Dental surgery may exacerbate ONJ in patients who develop ONJ while on bisphosphonate therapy. ■ Severe and occasionally incapacitating bone, joint, and/or muscle pain has been reported rarely. Onset of symptoms varied from one day to several months after beginning treatment with pamidronate. In most cases, pain resolves when pamidronate is discontinued; however, in some patients symptoms resolved slowly or persisted. ■ Atypical subtrochanteric and diaphyseal femoral fractures have been reported. May occur after minimal or no trauma. Risk may be increased in patients receiving concurrent glucocorticoids (e.g., dexamethasone, prednisone). ■ May be at risk for anemia, leukopenia, or thrombocytopenia; see Monitor. ■ Patients with a history of thyroid surgery may have relative hypoparathyroidism that may predispose them to hypocalcemia with pamidronate.
Osteolytic bone lesions of multiple myeloma and osteolytic bone metastases of breast cancer:
Patients being treated for multiple myeloma and bone metastases should have the dose withheld if renal function has deteriorated; see Dose Adjustments. Use is not recommended in patients with severe renal impairment being treated for bone metastases. See Monitor and Dose Adjustments. Limited information available on use in multiple myeloma patients with a CrCl less than 30 mL/min. In clinical trials, patients with a SCr above 3 mg/dL were excluded. ■ In the absence of hypercalcemia, patients with multiple myeloma or Paget’s disease of the bone or patients with predominantly lytic bone metastases who are at risk for calcium and vitamin D deficiency should be given oral calcium and vitamin D to reduce the risk of hypocalcemia.
Monitor:
Obtain baseline measurements of serum calcium (corrected for serum albumin), electrolytes, phosphate, magnesium, and creatinine and CBC with differential and hematocrit/hemoglobin. Monitor all closely as indicated by baseline results (may be daily). Serum phosphate levels will decrease and may require treatment. ■ Monitor renal function before each treatment; deterioration in renal function has been reported; see Precautions. ■ Monitor serum alkaline phosphatase during therapy for Paget’s disease. ■ Patients with cancer-related hypercalcemia are frequently dehydrated. Must be adequately hydrated orally and/or IV before treatment is initiated. Hydration with saline is preferred to facilitate renal excretion of calcium and to correct dehydration. A pretreatment urine output of 2 L/day is recommended. Maintain adequate hydration and urine output throughout treatment. ■ Avoid overhydration in patients with compromised cardiovascular status. Observe frequently for signs of fluid overload. Correct hypovolemia before using diuretics. ■ Monitor patients with pre-existing anemia, leukopenia, or thrombocytopenia very carefully during treatment and for the first 2 weeks following treatment. ■ Monitor for S/S of atypical femoral fractures that may occur with minimal or no trauma. Thigh or groin pain may be experienced weeks to months before a fracture appears. Fractures are often bilateral; examine both femurs.
Osteolytic bone lesions of multiple myeloma:
Adequately hydrate patients who have marked Bence-Jones proteinuria and dehydration before pamidronate infusion.
Patient education:
Regular visits and assessment of lab tests imperative. ■ Dietary restriction of calcium and vitamin D may be required. ■ Take only prescribed medications. ■ Report abdominal cramps, chills, confusion, fever, muscle spasms, sore throat, and/or any new medical problems promptly. ■ Report development of bone, joint, or muscle pain promptly. Onset of pain is variable. ■ Avoid pregnancy; use of nonhormonal birth control suggested.
Maternal/child:
Category D: should not be used during pregnancy. May cause fetal harm. Avoid pregnancy; use of birth control necessary during treatment and for an undetermined time after treatment; see prescribing information. ■ Discontinue breast-feeding. ■ Safety for use in pediatric patients not established.
Elderly:
Response similar to that seen in younger patients. ■ Use with caution based on age-related impaired organ function and concomitant disease or drug therapy; monitor renal function closely. See Dose Adjustments. ■ Monitor fluid and electrolyte status carefully to avoid overhydration or electrolyte imbalance. Use of lower fluid volume may be required; see Dilution.
Drug/lab interactions
Use caution when administered with other potentially nephrotoxic drugs (e.g., aminoglycosides [amikacin, tobramycin, gentamicin], cisplatin). ■ Concurrent administration with furosemide (Lasix) does not affect calcium-lowering action of pamidronate. ■ Does not interfere with any known primary cancer therapy. ■ Effects may be antagonized by calcium-containing preparations or vitamin D; avoid use. ■ Concurrent use with thalidomide (Thalomid) may increase risk of renal toxicity in patients with multiple myeloma.
Side effects
Average dose:
Abdominal pain, anemia, anorexia, bone pain, confusion and visual hallucinations (sometimes in conjunction with electrolyte imbalance), constipation, fever (mild and transient), generalized pain, hypertension, hypocalcemia (abdominal cramps, confusion, muscle spasms), infusion site reaction (e.g., induration and pain on palpation, redness, swelling), musculoskeletal pain (bone, joint, and/or muscle pain), pruritus, rash, renal toxicity, seizures, urinary tract infections, vomiting. Fluid overload, hypokalemia, hypomagnesemia, and hypophosphatemia occur frequently with use of concurrent fluid and diuretics. Rare instances of hypersensitivity reactions, including anaphylaxis, angioedema, dyspnea, and hypotension, have occurred. Osteonecrosis (primarily of the jaw) has been reported (see Precautions). Anemia, leukopenia, and thrombocytopenia may occur.
Overdose:
Occurs less frequently with lower dose range (30 to 60 mg). Fever (high), hypocalcemia, hypotension, leukopenia or lymphopenia (fever, chills, sore throat), transient taste perversion. Elevated BUN and CrCl levels and renal tubular necrosis may occur with excessive dose or rate of administration.
Post-marketing:
Adult respiratory distress syndrome (ARDS); atypical subtrochanteric and diaphyseal femoral fractures; bone, joint, and muscle pain (may be severe and incapacitating); conjunctivitis; focal segmental glomerulosclerosis (including the collapsing variant); glomerulonephropathies; hematuria; hypernatremia; influenza-like symptoms; interstitial lung disease; nephrotic syndrome; orbital inflammation; reactivation of herpes simplex and herpes zoster; renal tubular disorders; tubulointerstitial nephritis.
Antidote
Keep physician informed of side effects. Some may respond to symptomatic treatment. Magnesium, phosphorus, and potassium may require replacement if depletion too severe. If mild, all will probably return toward normal in 7 to 10 days. For asymptomatic or mild to moderate hypocalcemia (6.5 to 8 mg/100 mL [1 dL] corrected for serum albumin), short-term calcium therapy (e.g., calcium gluconate) may be indicated. Consider discontinuing pamidronate in patients who develop atypical fractures of the femur. Unknown if risk continues after stopping treatment. Discontinue drug for any symptoms of overdose. Monitor serum calcium and use vigorous IV hydration, with or without diuretics, for 2 to 3 days. Monitor intake and output to ensure adequacy and balance. Use short-term IV calcium therapy if indicated. High fever may respond to steroids. RBC transfusions may be required in anemia. Treat anaphylaxis and resuscitate as indicated.
Pancuronium bromide 
(pan-kyou-ROH-nee-um BRO-myd)
Neuromuscular blocking agent (nondepolarizing)
Anesthesia adjunct
pH 4
Usual dose
Adjunct to general anesthesia for adults and pediatric patients:
Must be individualized, depending on previous drugs administered and degree and length of muscle relaxation required. Must be used with adequate anesthesia and/or sedation and after unconsciousness induced. Succinylcholine must show signs of wearing off before pancuronium is given. 0.04 to 0.1 mg/kg of body weight initially. 0.01 mg/kg in increments as required to maintain muscle relaxation; usually 25- to 60-minute intervals.
Endotracheal intubation:
0.06 to 0.1 mg/kg.
Support of intubated mechanically ventilated or respiratory-controlled adult ICU patients (unlabeled):
IV bolus injection:
0.1 to 0.2 mg/kg every 1 to 3 hours.
Continuous infusion:
Begin with a loading dose of 0.03 to 0.1 mg/kg followed by a maintenance dose of 0.06 to 0.1 mg/kg/hr. A lower-end or reduced dose may be indicated if administered more than 5 minutes after the start of an inhalation agent, when steady-state has been achieved, or in patients with organ dysfunction (e.g., impaired renal function). Adjust dose according to clinical assessment of the patient’s response. Use of a peripheral nerve stimulator is recommended.
Neonatal dose
Adjunct to general anesthesia:
Extreme sensitivity to pancuronium exists during the first month of life. Begin with a test dose of 0.02 mg/kg and assess responsiveness. See Maternal/Child.
Dose adjustments
See Drug/Lab Interactions; marked reduction of pancuronium dose may be required. ■ A higher total dose may be required in biliary or hepatic disease, but onset is slower and neuromuscular block is prolonged. ■ Clearance decreased and half-life increased in renal insufficiency. One source recommends administering 50% of a dose if CrCl is between 10 and 50 mL/min; do not use if CrCl is less than 10 mL/min.
Dilution
May be given undiluted or diluted in D5W, D5/1/2NS, D5NS, NS, or LR for use as an infusion.
Storage:
Best if stored in refrigerator. Will maintain potency at room temperature for up to 6 months.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Ciprofloxacin (Cipro IV), verapamil.
Y-site:
Aminophylline, cefazolin (Ancef), cefuroxime (Zinacef), dexmedetomidine (Precedex), dobutamine, dopamine, epinephrine (Adrenalin), esmolol (Brevibloc), etomidate (Amidate), fenoldopam (Corlopam), fentanyl, fluconazole (Diflucan), gentamicin, heparin, hetastarch in electrolytes (Hextend), hydrocortisone sodium succinate (Solu-Cortef), isoproterenol (Isuprel), levofloxacin (Levaquin), lorazepam (Ativan), midazolam (Versed), milrinone (Primacor), morphine, nitroglycerin IV, nitroprusside sodium, propofol (Diprivan), ranitidine (Zantac), sulfamethoxazole/trimethoprim, vancomycin.
Rate of administration
Adjunct to general anesthesia:
A single dose over 60 to 90 seconds.
Mechanical ventilation support in ICU:
See Usual Dose for specific rates and criteria.
Actions
A skeletal muscle relaxant. Causes paralysis by interfering with neural transmission at the myoneural junction. Onset of action is dose dependent. Peak effect occurs in 3 to 4 minutes and lasts 30 to 45 minutes. It may take another 30 minutes or up to several hours before complete recovery occurs. Excreted in the urine.
Indications and uses
Adjunctive to general anesthesia to facilitate endotracheal intubation and to relax skeletal muscles during surgery or mechanical ventilation.
Unlabeled uses:
Support of intubated, mechanically ventilated, or respiratory-controlled patients in ICU.
Contraindications
Known hypersensitivity to pancuronium or bromides; first trimester of pregnancy.
Precautions
Usually administered by or under the direct observation of the anesthesiologist. Appropriate emergency drugs and equipment for monitoring the patient and responding to any medical emergency must be readily available. ■ Severe anaphylactic reactions have been reported with neuromuscular blocking agents; some have been fatal. Use caution in patients who have had an anaphylactic reaction to another neuromuscular blocking agent (depolarizing or nondepolarizing); cross-reactivity has occurred. ■ Repeated doses may produce a cumulative effect. ■ Impaired pulmonary function or respiratory deficiencies can cause critical reactions. ■ Use caution in impaired liver or kidney function, in patients with tachycardia, and in any patient who might develop adverse effects from an increase in HR. ■ Myasthenia gravis increases sensitivity to drug. ■ Long-term use (i.e., intensive care) may result in prolonged paralysis or skeletal muscle weakness. ■ Acid-base and/or electrolyte imbalance, debilitation, hypoxic episodes, and/or the use of other drugs (e.g., broad-spectrum antibiotics, narcotics, steroids) may prolong the effects of pancuronium.
Monitor:
All uses:
This drug produces apnea. Controlled artificial ventilation with oxygen must be continuous and under direct observation at all times. Maintain a patent airway. ■ Use a peripheral nerve stimulator to monitor response to pancuronium and avoid overdose. ■ Patient may be conscious and completely unable to communicate by any means. Pancuronium has no analgesic or sedative properties. ■ Action is altered by dehydration, electrolyte imbalance, body temperatures, and acid-base imbalance. ■ Hyperkalemia may cause cardiac arrhythmias and increased paralysis. ■ See Precautions and Drug/Lab Interactions.
Mechanical ventilation support in ICU:
Physical therapy is recommended to prevent muscular weakness, atrophy, and joint contracture. Muscular weakness may first be noticed during attempts to wean patients from the ventilator.
Maternal/child:
Category C: unknown potential hazards to fetus. Benefits must outweigh risks. Not recommended in first trimester. ■ Has caused rare severe skeletal muscle weakness in neonates undergoing mechanical ventilation.
Elderly:
Delay in onset time may be caused by slower circulation time in cardiovascular disease, old age, or edematous states; allow more time for drug to achieve maximum effect.
Drug/lab interactions
May cause severe arrhythmias with inhalant anesthetics (e.g., enflurane, halothane) and in patients on chronic tricyclic antidepressant therapy (e.g., amitriptyline [Elavil]). ■ Potentiated by hypokalemia, some carcinomas, many antibiotics (e.g., aminoglycosides [kanamycin, gentamicin], bacitracin, colistin [Coly-Mycin S], colistimethate [Coly-Mycin M], polymyxin-B [Aerosporin], tetracyclines, piperacillin), calcium salts, CO2, diuretics, diazepam (Valium) and other muscle relaxants, digoxin, magnesium sulfate, quinidine, morphine, lidocaine, meperidine, propranolol (Inderal), succinylcholine, and others. May need to reduce dose of pancuronium; use with caution. ■ Recurrent paralysis may occur with quinidine. ■ Effects may be decreased by acetylcholine, aminophylline, anticholinesterases, azathioprine, carbamazepine, and potassium. ■ Succinylcholine must show signs of wearing off before pancuronium is given. Use caution.
Side effects
Increased HR, decrease in mean arterial pressure, prolonged action resulting in respiratory insufficiency or apnea and tachycardia. Airway closure caused by relaxation of epiglottis, pharynx, and tongue muscles. Hypersensitivity reactions are possible. Anaphylaxis, histamine release (e.g., bronchospasm, vasodilation, hypotension, cutaneous flushing), and shock may occur. Muscular weakness and atrophy may occur with long-term use (1 to 3 weeks).
Antidote
All side effects are medical emergencies. Treat symptomatically. Controlled artificial ventilation must be continuous. Pyridostigmine (Regonol) or neostigmine given with atropine will probably reverse the muscle relaxation. Not effective in all situations; may aggravate severe overdose. Resuscitate as necessary.
Panitumumab 
(pan-i-TUE-moo-mab)
Vectibix
Antineoplastic
Immunosuppressant
Monoclonal antibody
pH 5.6 to 6
Usual dose
Pre-testing required:
Before initiating treatment, assess RAS mutational status in colorectal tumors and confirm the absence of a RAS mutation; see Precautions. See prescribing information for access to FDA-approved tests.
Panitumumab:
6 mg/kg as an infusion once every 14 days.
Dose adjustments
Upon the first occurrence of a Grade 3 (NCI-CTC/CTCAE) dermatologic reaction, withhold 1 to 2 doses of panitumumab. If the reaction improves to less than Grade 3, reinitiate panitumumab at the original dose. ■ Upon the second occurrence of a Grade 3 (NCI-CTC/CTCAE) dermatologic reaction, withhold 1 to 2 doses of panitumumab. If the reaction improves to less than Grade 3, reinitiate panitumumab at 80% of the original dose. ■ Upon the third occurrence of a Grade 3 (NCI-CTC/CTCAE) dermatologic reaction, withhold 1 to 2 doses of panitumumab. If the reaction improves to less than Grade 3, reinitiate panitumumab at 60% of the original dose. ■ Upon the fourth occurrence of a Grade 3 (NCI-CTC/CTCAE) dermatologic reaction, permanently discontinue panitumumab. ■ Permanently discontinue panitumumab following the occurrence of a Grade 4 dermatologic reaction or for a Grade 3 dermatologic reaction that does not recover after withholding 1 to 2 doses.
Dilution
Solution may contain a small amount of visible, translucent-to-white, amorphous, and proteinaceous panitumumab particulates (will be removed by in-line filter). Do not use if solution is discolored. Do not shake. Withdraw calculated dose from vial. Dilute to a total volume of 100 mL with NS. Doses higher than 1,000 mg should be diluted to a volume of 150 mL. (Withdraw a volume of NS equal to the volume of the calculated dose from the infusion bag.) Final concentration should not exceed 10 mg/mL. Mix diluted solution by gentle inversion. Do not shake.
Filters:
Must be administered through a low–protein binding, 0.2- or 0.22-micron, in-line filter.
Storage:
Store unopened vials in refrigerator (2° to 8° C [36° to 46° F]). Protect from direct sunlight. Do not freeze. Diluted solutions should be used within 6 hours of preparation if stored at RT and within 24 hours if stored in refrigerator. Do not freeze. Single-dose vial; discard any unused product after entry into vial.
Compatibility
Manufacturer states, “Should not be mixed with, or administered as an infusion with, other medicinal products. No other medications should be added to solutions containing panitumumab.”
Rate of administration
Flush line before and after panitumumab administration with NS. Do not administer as an IV push or bolus. Administer with an IV infusion pump using a low–protein binding, 0.2- or 0.22-micron, in-line filter.
A single dose equally distributed over 60 minutes. If the first infusion is tolerated, administer subsequent infusions over 30 to 60 minutes. Doses over 1,000 mg should be administered over 90 minutes. Reduce rate of infusion by 50% in patients experiencing a Grade 1 or 2 infusion reaction.
Actions
A genetically engineered recombinant, human IgG2 kappa monoclonal antibody that binds specifically to the human epidermal growth factor receptor (EGFR). EGFR is a transmembrane glycoprotein that is expressed in many normal epithelial tissues, including skin and hair follicles. Overexpression of EGFR is detected in many human cancers, including those of the colon and rectum. Interaction of EGFR with its normal ligands leads to a series of reactions that regulate the transcription of molecules involved with cellular growth and survival, motility, proliferation, and transformation. Panitumumab binds to EGFR on both normal and tumor cells, inhibiting the binding of normal ligands to EGFR. This competitive binding results in inhibition of cell growth, induction of apoptosis, decreased proinflammatory cytokine and vascular growth factor production, and internalization of EGFR. The end result is inhibition of growth and survival of selected human tumor cells that express EGFR. Half-life is approximately 7.5 days (range 3.6 to 10.9 days).
Indications and uses
Treatment of patients with wild-type KRAS (exon 2 in codons 12 or 13) metastatic colorectal cancer (mCRC) as determined by an FDA-approved test for this use. May be used as first-line therapy in combination with FOLFOX (5-fluorouracil [5-FU], leucovorin, and oxaliplatin [Eloxatin]) or as monotherapy following disease progression after prior treatment with fluoropyrimidine (fluorouracil [5-FU])-, oxaliplatin-, and irinotecan (Camptosar)-containing chemotherapy.
Limitation of use:
Not indicated for the treatment of patients with RAS mutant mCRC or for patients whose RAS mutation status is unknown.
Contraindications
None known. See Precautions.
Precautions
Panitumumab should be used only for treatment of patients with KRAS wild-type mCRC as determined by an FDA-approved test. In studies, patients with KRAS-mutant mCRC tumors receiving panitumumab in combination with FOLFOX experienced some overall survival compared with patients receiving FOLFOX alone. Perform the assessment for KRAS mutational status in colorectal cancer laboratories with demonstrated proficiency in the technology required for testing. ■ Dermatologic toxicities were reported in 90% of patients and were severe (NCI-CTC/CTCAE Grade 3 or higher) in 15% of patients. Manifestations included dermatitis acneiform, dry skin, erythema, paronychia, pruritus, rash, skin exfoliation, and skin fissures. Severe dermatologic toxicities were complicated by infection. Life-threatening and fatal infectious complications, including necrotizing fasciitis, abscesses requiring incision and drainage, and sepsis, have been observed. Life-threatening and fatal bullous mucocutaneous disease with blisters, erosions, and skin sloughing has also been reported. It is unclear whether these reactions were directly related to EGFR inhibition or to idiosyncratic immune-related effects (e.g., Stevens-Johnson syndrome or toxic epidermal necrolysis). See Dose Adjustments and Antidote. ■ Sunlight may exacerbate dermatologic toxicity. Protection from sun is advised; see Patient Education. ■ Infusion reactions, some severe, have been reported. Fatal reactions have been reported. Administer in a facility with adequate emergency medical equipment and medications for treating these reactions and for responding to any medical emergency; see Rate of Administration and Antidote. ■ Increased mortality and toxicity were seen in studies in which panitumumab was administered in combination with bevacizumab (Avastin) and chemotherapy; see manufacturer’s prescribing information. ■ Electrolyte abnormalities have been reported; see Monitor. ■ Fatal and nonfatal cases of interstitial lung disease (ILD) and pulmonary fibrosis have been reported; see Monitor. In patients with a history of interstitial pneumonitis or pulmonary fibrosis or with evidence of interstitial pneumonitis or pulmonary fibrosis, the benefit of therapy versus the risk of pulmonary complications must be considered. ■ Severe diarrhea and dehydration leading to acute renal failure and other complications have been observed in patients treated with panitumumab in combination with chemotherapy. ■ Keratitis and ulcerative keratitis, known risk factors for corneal perforation, have been reported. ■ A protein substance. Potential for immunogenicity exists. Anti-panitumumab antibodies have been detected in a small number of patients. No evidence of altered safety profiles has been confirmed.
Monitor:
Obtain baseline electrolytes and monitor periodically during and for 8 weeks after completion of therapy. Hypomagnesemia, hypocalcemia, and hypokalemia have been reported. Oral or IV electrolyte replacement may be required. ■ Monitor hydration status. ■ Monitor vital signs before, during (as needed), and at the completion of the infusion. ■ Monitor for S/S of hypersensitivity or infusion-related reactions. Severe reactions may include anaphylaxis, bronchospasm, chills, dyspnea, fever, and hypotension; see Side Effects. Mild to moderate infusion reactions may respond to a reduction in the rate of infusion; see Rate of Administration. Utility of premedication to minimize or prevent infusion-related reactions has not been determined. ■ Monitor skin integrity. Monitor patients who develop dermatologic or soft tissue toxicities for the development of inflammatory or infectious sequelae. ■ Monitor lung function. Interrupt therapy in the event of acute onset or worsening of pulmonary symptoms. Discontinue therapy if ILD is confirmed. ■ Monitor for evidence of keratitis or ulcerative keratitis.
Patient education:
Avoid pregnancy. Nonhormonal birth control recommended for both men and women during and for 6 months following completion of therapy. ■ Review side effects, including dermatologic toxicity, infusion reactions, pulmonary fibrosis, and potential for fetal harm. ■ Report skin or ocular changes, cough, dehydration, diarrhea, dyspnea, or infusion-related reactions promptly. ■ Limit sun exposure during and for 2 months after the last dose of panitumumab. Use of sunscreen and hats recommended. ■ Compliance with periodic lab work required.
Maternal/child:
Category C: safety for use in pregnancy has not been established. EGFR is thought to be involved in prenatal development and may be essential for normal organogenesis, proliferation, and differentiation in the developing embryo. Human IgG crosses the placental barrier. Therefore it is possible that panitumumab may be transmitted from the mother to the developing fetus. Appropriate contraceptive measures must be used during treatment with panitumumab; see Patient Education. ■ Women who become pregnant during panitumumab therapy are encouraged to enroll in Amgen’s Pregnancy Surveillance Program. ■ Discontinue breast-feeding during and for 2 months after the completion of therapy. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
When used as monotherapy, specific differences in safety and effectiveness compared with younger adults not noted. An increased incidence of serious adverse events and an increased incidence of serious diarrhea were seen in patients over the age of 65 when given panitumumab in combination with FOLFOX.
Drug/lab interactions
Side effects
The most commonly reported side effects of panitumumab as monotherapy are diarrhea, fatigue, nausea, paronychia, and skin rash with variable presentations. The most common serious reactions, as well as the reactions that lead most often to discontinuation of therapy, were general physical health deterioration and intestinal obstruction. Side effects occurring in 5% or more of patients receiving panitumumab as monotherapy included acne, acneiform dermatitis, cough, dry skin, dyspnea, erythema, exfoliative rash, mucosal inflammation, nail disorders, rash, skin exfoliation, skin fissures, skin ulcer, stomatitis, and vomiting. The most commonly reported side effects when panitumumab was used in combination with chemotherapy were acneiform dermatitis, anorexia, asthenia, diarrhea, dry skin, hypokalemia, hypomagnesemia, mucosal inflammation, paronychia, pruritus, rash, and stomatitis. The most common serious reactions were diarrhea and dehydration. The most commonly reported side effects leading to discontinuation were acneiform dermatitis, diarrhea, fatigue, hypersensitivity, paresthesia, and rash. Side effects occurring in 5% or more of patients receiving panitumumab in combination with chemotherapy included acne, alopecia, conjunctivitis, dehydration, epistaxis, erythema, nail disorders, palmar-plantar erythrodysesthesia syndrome, skin fissures, and weight decrease. Other serious but less frequently reported side effects as outlined in Precautions included acute renal failure, infusion reactions, interstitial lung disease/pulmonary fibrosis, and ocular toxicity.
Post-marketing:
Angioedema, infusion reactions (fatal), keratitis/ulcerative keratitis, life-threatening and fatal bullous mucocutaneous disease, and skin necrosis.
Antidote
Notify physician of any side effects; most will be treated symptomatically. Replace electrolytes parenterally or orally as indicated. Reduce infusion rate by 50% if a mild or moderate (Grade 1 or 2) infusion reaction occurs. Discontinue panitumumab for a serious infusion reaction (Grade 3 or 4). Depending on the severity and/or persistence of the reaction, discontinue permanently. Hold or discontinue panitumumab if dermatologic or soft tissue toxicities Grade 3 or higher occur, if they are considered intolerable, or if severe or life-threatening inflammatory or infectious complications occur. See Dose Adjustments for criteria to resume or permanently discontinue treatment. Discontinue if ILD is confirmed. Interrupt or discontinue panitumumab infusion for acute or worsening keratitis. Treat hypersensitivity or infusion reaction as indicated (e.g., oxygen, antihistamines [e.g., diphenhydramine (Benadryl)], epinephrine [Adrenalin], corticosteroids, vasopressors [e.g., dopamine], ventilation equipment, and/or fluids). Resuscitate as necessary.
Pantoprazole sodium
(pan-TOH-prah-zohl SO-dee-um)
Protonix IV
Proton pump inhibitor (Gastric acid inhibitor)
pH 9 to 10.5
Usual dose
Given as an alternative to continued oral therapy. Resume oral therapy as soon as practical.
Treatment of gastroesophageal reflux disease (GERD):
40 mg as an infusion once daily for 7 to 10 days.
Treatment of Zollinger-Ellison syndrome (ZES):
80 mg as an infusion every 12 hours. Adjust to patient needs based on acid output measurements. If an increased dose is required, 80 mg every 8 hours is expected to maintain gastric acid output at less than 10 mEq/hr. Daily doses in excess of 240 mg or for more than 6 days have not been studied.
Recurrent gastrointestinal bleeding, prophylaxis (unlabeled):
80 mg as an IV injection followed by a continuous infusion of 8 mg/hr for 72 hours.
Dose adjustments
No dose adjustments are necessary based on race or gender; in the elderly; in patients with mild, moderate, or severe renal insufficiency; in patients on hemodialysis; or in patients with mild to severe impaired hepatic function. Doses higher than 40 mg/day have not been studied in patients with hepatic impairment.
Dilution
40-mg dose:
Reconstitute each single dose with 10 mL NS. May be further diluted with 100 mL D5W, NS, or LR to achieve a final concentration of approximately 0.4 mg/mL for infusion.
80-mg dose:
Combining of two 40-mg vials is required; reconstitute each 40-mg vial with 10 mL NS. This total 80-mg dose may be further diluted with 80 mL D5W, NS, or LR (a total volume of 100 mL) to achieve a final concentration of approximately 0.8 mg/mL for infusion.
Filters:
No longer required.
Storage:
Store unopened vials at CRT. Protect from light. Do not freeze reconstituted product. Vials reconstituted for the 2-minute infusion may be stored for up to 24 hours at RT. Vials reconstituted for the 15-minute infusion may be stored for up to 6 hours at RT prior to further dilution. Fully diluted solution may then be stored at RT but must be used within 24 hours of initial reconstitution. Protection from light is not required for reconstituted or fully diluted solutions.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Administer through a dedicated line or through a Y-site. Should not be simultaneously administered through the same line with other intravenous solutions.” See Rate of Administration. Manufacturer lists as incompatible at the Y-site with midazolam (Versed) and states, “May not be compatible with products containing zinc.” Discontinue if discoloration or precipitation occurs with any drug at the Y-site.
One source suggests the following compatibilities:
Y-site:
Ampicillin, anidulafungin (Eraxis), caspofungin (Cancidas), cefazolin (Ancef), ceftaroline (Teflaro), ceftriaxone (Rocephin), dimenhydrinate, dopamine, doripenem (Doribax), epinephrine (Adrenalin), furosemide (Lasix), insulin (regular), morphine, nitroglycerin IV, norepinephrine (Levophed), octreotide (Sandostatin), potassium chloride (KCl), telavancin (Vibativ), vasopressin.
Rate of administration
Flush IV line before and after administration of pantoprazole with compatible infusion solutions (D5W, NS, or LR).
Concentration determines the rate of administration.
40 mg/10 ml or 80 mg/20 mL:
As an injection evenly distributed over at least 2 minutes.
40 mg/100 ml or 80 mg/100 mL:
As an infusion evenly distributed over at least 15 minutes (approximately 7 mL/min).
Actions
A proton pump inhibitor (PPI) that suppresses the final step in gastric acid production. Acts at the secretory surface of the gastric parietal cell. Inhibits both basal and stimulated gastric acid secretion irrespective of the stimulus. Does not accumulate and pharmacokinetics are not altered with multiple daily dosing. Onset of antisecretory activity is within 15 to 30 minutes. Half-life is approximately 1 hour. However, duration of antisecretory effect persists longer than 24 hours. Distributed mainly in extracellular fluid. Highly bound by serum protein (primarily albumin). Extensively metabolized in the liver through the cytochrome P450 system (primarily through CYP2C19 and, to a lesser extent, through CYP3A4). Metabolism is independent of the route of administration (intravenous or oral). Primarily excreted in urine with some excretion in feces. Secreted in breast milk.
Indications and uses
Short-term treatment (7 to 10 days) of gastroesophageal reflux disease (GERD) with a history of erosive esophagitis, as an alternative to oral therapy. ■ Treatment of pathologic hypersecretion associated with Zollinger-Ellison syndrome or other pathologic hypersecretory conditions.
Unlabeled uses:
Prophylaxis against recurrent GI bleed (peptic ulcer bleed).
Contraindications
Known hypersensitivity to pantoprazole (Protonix), other substituted benzimidazoles (e.g., esomeprazole [Nexium], omeprazole [Prilosec]), or any component of the formulation.
Precautions
For IV use only; do not give IM or SC. ■ Gastric malignancy may be present even though patient’s symptoms improve with pantoprazole therapy. ■ Formulation contains edetate disodium, which can chelate zinc; see Monitor. ■ Should be discontinued as soon as the patient is able to resume treatment with pantoprazole delayed-release tablets or oral suspension. ■ Data on safe and effective dosing for other conditions (including life-threatening upper GI bleeds) not available. 40 mg IV of pantoprazole daily does not raise gastric pH levels sufficiently to treat such life-threatening conditions. ■ Hypersensitivity reactions, including anaphylaxis and severe skin reactions (e.g., erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis), have been reported. ■ May be associated with an increased risk for osteoporosis-related fractures of the hip, wrist, or spine. Risk increased in patients receiving high-dose (multiple daily doses) and long-term therapy (a year or longer). Use lowest dose and shortest duration of therapy appropriate for the condition being treated. ■ In patients treated with proton pump inhibitors (PPIs) for at least 3 months and, in most cases, after a year of treatment, hypomagnesemia (symptomatic and asymptomatic) has been reported. Serious adverse events, including arrhythmias, seizures, and tetany, have occurred. Discontinuation of the PPI and magnesium replacement have been required. ■ May be associated with an increased risk of Clostridium difficile–associated diarrhea (CDAD), especially in hospitalized patients. Consider in patients who develop diarrhea that does not improve. ■ Acute interstitial nephritis has been observed and is generally attributed to an idiopathic hypersensitivity reaction. May occur at any time during therapy. Discontinue pantoprazole if it occurs. ■ Use caution in transplant patients receiving mycophenolate mofetil (CellCept). ■ See Drug/Lab Interactions.
Monitor:
Observe for S/S of a hypersensitivity reaction and/or severe skin reaction (e.g., acute interstitial nephritis, anaphylactic shock, anaphylaxis, angioedema, bronchospasm, urticaria). ■ Monitor vital signs and pain levels. ■ Concomitant use of antacids may be indicated. ■ In hypersecretory states, acid output measurements may be indicated to guide dose adjustment. ■ Monitor injection site. Thrombophlebitis has been reported. ■ Zinc supplementation may be indicated in patients who are prone to zinc deficiency; see Precautions. ■ Monitoring of magnesium levels may be indicated with prolonged PPI therapy; see Precautions and Drug/Lab Interactions. ■ Change to oral dosing when appropriate. ■ See Drug/Lab Interactions.
Patient education:
Review prescription and nonprescription drugs with your physician. ■ Oral route preferred.
Maternal/child:
Category B: use during pregnancy only when clearly needed. ■ Has potential for serious harm to nursing infants; discontinue breast-feeding. ■ Safety and effectiveness for pediatric patients under 18 years of age not established.
Elderly:
Safety and effectiveness similar to that of younger patients.
Drug/lab interactions
Because of profound and long-lasting inhibition of gastric acid secretion, pantoprazole may interfere with the absorption of drugs in which gastric pH is an important determinant of their bioavailability. Absorption of atazanavir (Reyataz), erlotinib (Tarceva), iron salts (ferrous sulfate), ketoconazole (Nizoral), and mycophenolate mofetil (CellCept) can decrease; other drugs (e.g., digoxin [Lanoxin]) can increase. Coadministration of mycophenolate mofetil (MMF) and omeprazole (Prilosec) in transplant patients receiving MMF reduces the exposure to MMF’s active metabolite, mycophenolic acid; see Precautions. ■ Increases in INR and PT have been reported when administered concurrently with warfarin (Coumadin); monitoring of INR and PT indicated. ■ Concurrent use with atazanavir (Reyataz) or nelfinavir (Viracept) is not recommended. Coadministration of proton pump inhibitors (e.g., esomeprazole [Nexium], pantoprazole [Protonix]) results in a significant reduction in atazanavir or nelfinavir plasma concentrations, thereby inhibiting atazanavir’s therapeutic effect and increasing development of drug resistance. ■ Long-term use may increase the risk of digoxin toxicity secondary to hypomagnesemia. ■ Patients on long-term therapy receiving concomitant diuretic therapy may be at increased risk for hypomagnesemia. ■ Concomitant use of proton pump inhibitors with high-dose methotrexate may elevate and prolong serum levels of methotrexate and/or its metabolite. May lead to methotrexate toxicity; consider withdrawal of pantoprazole. ■ Concomitant administration of pantoprazole and clopidogrel (Plavix) had no clinically significant effect on clopidogrel-induced platelet aggregation. No dose adjustment of clopidogrel is necessary when administered with approved doses of pantoprazole. ■ Studies suggest that with concurrent use the metabolism and serum concentrations of pantoprazole are not significantly altered by other drugs metabolized by the cytochrome P450 system (e.g., clarithromycin [Biaxin], diazepam [Valium], diclofenac [Apo-Diclo], metoprolol [Lopressor], midazolam [Versed], naproxen [Aleve], nifedipine [ProCardia XL], phenytoin [Dilantin], piroxicam [Feldene], theophylline). ■ Studies also suggest that concurrent use with pantoprazole has not been found to significantly alter the metabolism and serum concentrations of the above agents as well as other drugs metabolized by a similar process (e.g., carbamazepine [Tegretol], cisapride [Propulsid], oral contraceptives). Concurrent administration should not require dose adjustment of either drug. ■ Amoxicillin, caffeine, digoxin (Lanoxin), ethanol, glyburide (DiaBeta), and metronidazole (Flagyl IV) had no clinically relevant interactions during clinical studies. ■ May cause false-positive urine screening test for tetrahydrocannabinol (THC).
Side effects
The most frequently occurring side effects are abdominal pain, arthralgias, diarrhea, dizziness, flatulence, headache, nausea, and vomiting. Abscess, blurred vision, chest pain, confusion, dyspnea, gastroenteritis, hemorrhage, hyperglycemia, hypokinesia, increased salivation, injection site reactions including thrombophlebitis, pruritus, rash, speech disorder, tinnitus, transient elevations of serum transaminase, urinary tract infection, and vertigo are reported. Numerous other side effects have been associated with oral pantoprazole.
Post-marketing:
Agranulocytosis, anterior ischemic optic neuropathy, asthenia, bone fracture, CDAD, elevated creatine phosphokinase (CPK), fatigue, hallucinations, hepatocellular damage leading to jaundice and hepatic failure, hypersensitivity reactions (e.g., anaphylaxis, angioedema), hypomagnesemia, hyponatremia, insomnia, interstitial nephritis, malaise, pancreatitis, pancytopenia, rhabdomyolysis, severe dermatologic reactions (e.g., erythema multiforme, Stevens-Johnson syndrome, taste disorders, toxic epidermal necrolysis [some fatal]), somnolence, weight gain.
Antidote
Keep physician informed of all side effects. May be treated symptomatically. Discontinue and initiate appropriate treatment if S/S associated with post-marketing reports occur; see Side Effects. Adverse effects from overdose (up to 240 mg/day in healthy subjects) did not occur during clinical trials. Not removed by hemodialysis.
Papaverine hydrochloride
(pah-PAV-er-een hy-droh-KLOR-eyed)
Vasodilator (peripheral)
pH 3 to 4.5
Usual dose
1 to 4 mL (30 to 120 mg) every 3 hours as indicated. Second dose may be given in 10 minutes only when treating extrasystoles.
Pediatric dose
1.5 mg/kg of body weight every 6 hours; see Maternal/Child.
Dose adjustments
Dilution
May be given undiluted or may be diluted in an equal amount of SWFI. Usually not added to IV solutions. May be given through Y-tube or three-way stopcock of infusion set.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Will form a precipitate with LR.
One source suggests the following compatibilities:
Additive:
Theophylline.
Rate of administration
1 mL (30 mg) or fraction thereof over 2 minutes. Rapid IV injection may cause death.
Actions
A nonnarcotic opium alkaloid, it is a direct smooth muscle relaxant and antispasmodic. Relaxation is noted in vascular system and bronchial musculature and in GI, biliary, and urinary tracts. More effective on muscle in spasm, it has an affinity for the smooth muscle of blood vessels. Affects cardiac muscle to depress conduction and increase refractory period. Improved circulation and muscle relaxation decrease pain. Metabolized in the liver and excreted in the urine.
Indications and uses
Vascular spasm associated with an acute myocardial infarction. ■ Peripheral or pulmonary embolism. ■ Peripheral vascular disease and cerebral angiospastic states. ■ Visceral spasm of ureteral, biliary, or GI colic. ■ Angina pectoris.
Contraindications
Complete AV heart block.
Precautions
Rarely used; active therapeutic value is questioned. ■ Rapid IV injection may cause death. ■ IM injection is preferred. ■ Use with caution in glaucoma and impaired liver function. ■ Large doses can depress AV and intraventricular conduction, resulting in arrhythmias.
Monitor:
Observe patient continuously; monitor vital signs. ■ See Drug/Lab Interactions.
Patient education:
Avoid alcohol and other CNS depressants. ■ May cause dizziness and drowsiness; request assistance for ambulation. ■ Use caution in any task requiring alertness.
Maternal/child:
Category C: safety for use in pregnancy and breast-feeding and in pediatric patients not established.
Elderly:
Risk of hypothermia may be increased.
Drug/lab interactions
May be used with narcotics if the relaxant effect is not adequate to relieve discomfort. Narcotic dosage should be reduced. ■ Antagonizes effects of levodopa.
Side effects
Blurred or double vision, diaphoresis, discomfort (generalized), flushing, hypertension (slight), hypotension, respiratory depth increase, scleral jaundice, sedation, tachycardia.
Major:
Respiratory depression, seizures, ventricular ectopic rhythms, sudden death.
Antidote
Notify the physician of any minor side effects. If minor symptoms progress or any major side effect appears, discontinue the drug immediately and notify the physician. Treatment of toxicity will be symptomatic and supportive. Consider diazepam (Valium) or phenytoin (Dilantin) for convulsions. Anesthesia with thiopental and paralysis with a neuromuscular blocking agent may be required. Use dopamine for hypotension. Calcium gluconate may reduce toxic cardiovascular effects. Monitor ECG. Resuscitate as necessary.
Paricalcitol
(pair-ee-KAL-sih-tohl)
Zemplar
Vitamin D analog
Usual dose
The currently accepted target range for intact parathyroid hormone (iPTH) in chronic kidney disease (CKD) Stage 5 patients is no more than 1.5 to 3 times the nonuremic upper limit of normal.
Recommended initial dose is 0.04 to 0.1 mcg/kg administered no more frequently than every other day at any time during dialysis. Doses as high as 0.24 mcg/kg have been safely administered.
Information supplied by the manufacturer suggests that the relative dosing of paricalcitol to calcitriol is 4:1. When converting a patient from calcitriol to paricalcitol, the initial dose of paricalcitol should be four times greater than the patient’s dose of calcitriol.
Pediatric dose
Dose recommendations are based on severity of disease in addition to body weight. A dose is administered at any time during dialysis.
Pediatric patients 5 to 19 years of age (unlabeled):
Doses used in clinical trials were:
Baseline iPTH level less than 500 pg/mL:
0.04 mcg/kg 3 times a week.
Baseline iPTH level equal to or greater than 500 pg/mL:
0.08 mcg/kg 3 times a week.
See Dose Adjustments.
Dose adjustments
Adults:
Adjust dosing based on patient response. If a satisfactory response is not observed, dose may be increased by 2 to 4 mcg at 2- to 4-week intervals. Monitor serum calcium, phosphorus, and calcium × phosphorus product (Ca × P) frequently during any dose adjustment period. See Monitor.
Pediatric patients:
Adjust dose in 0.04-mcg/kg increments based on the levels of serum iPTH, calcium, and Ca × P (from clinical trials).
Adults and pediatric patients:
Reduce dose or interrupt therapy if elevated calcium level or a Ca × P product of greater than 75 is noted. Reinitiate therapy at a lower dose when parameters have normalized. ■ Paricalcitol dose may need to be reduced as parathyroid hormone (PTH) levels decrease in response to therapy. The currently accepted target range for intact parathyroid hormone (iPTH) in patients with chronic kidney disease (CKD) is no more than 1.5 to 3 times the nonuremic upper limit of normal. Incremental dosing must be individualized and commensurate with PTH, serum calcium, and phosphorus levels. The following chart is a suggested approach to dose titration.
| Paricalcitol Suggested Dosing Guidelines | |
| PTH Level | Paricalcitol Dose |
| The same or increasing | Increase |
| Decreasing by <30% | Increase |
| Decreasing by >30%, <60% | Maintain |
| Decreasing by >60% | Decrease |
| 11/2 to 3 times the upper limit of normal | Maintain |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


