23 Neuromuscular blocking agents
Action Potential: The passage of an electric impulse at any point on the nerve fiber where the inside becomes positive and the outside becomes negative; also referred to as action current.
Anticholinesterase: A drug that inhibits or inactivates the action of acetylcholinesterase.
Antimuscarinic (Anticholinergic): A drug that blocks the effects of acetylcholine receptors and results in the inhibition of the transmission of parasympathetic nerve impulses.
Clinical Duration: In reference to the use of neuromuscular blocking agents, the time from the administration of the drug to 25% recovery of the train-of-four twitch response.
Defasciculation: A result of the administration of a subclinical dose of a nondepolarizing skeletal muscle relaxant for prevention of the skeletal muscle twitches that occur after the administration of the depolarizing skeletal muscle relaxant succinylcholine.
Depolarizing Skeletal Muscle Relaxant: A skeletal muscle relaxant that after administration produces skeletal muscle twitches by stimulating the nicotinic receptors on the neuromuscular end plate and remaining on the end plate for 3 to 5 minutes, which leads to muscle paralysis. Skeletal muscle function returns as the pseudocholinesterase metabolizes the succinylcholine, usually between 3 and 5 minutes.
End Plate Potential (EPP): One action potential at the myoneural junction.
Excitation-Contraction (E-C) Coupling: The entire process of muscle contraction, starting with the electric and then the chemical stimulus to the process of the release of calcium in the sarcoplasmic reticulum, which causes the muscle fibers (actin and myosin) to slide and thus contract.
Extraocular Muscles: The six sets of muscles that control the movement of the eyeball.
Fasciculations: Skeletal muscle twitches.
Muscarinic: Subset receptors of the parasympathetic nervous system.
Myopathy: An abnormal condition of skeletal muscle characterized by muscle weakness and wasting.
Neurohumoral Transmission: Combined electric and chemical transmission of an impulse.
Nicotinic: Subset of the parasympathetic nervous system.
Nondepolarizing Agents: Drugs that cause paralysis of skeletal muscle by blocking neural muscular transmission at the myoneural junction.
Onset Time: In reference to the use of neuromuscular blocking agents, the time from the administration of the drug to maximum effect.
Pseudocholinesterase: An enzyme that acts like cholinesterase and metabolizes acetylcholine.
Recovery Index: In reference to the use of neuromuscular blocking agents, the time from the train-of-four twitch index of 25% to 75% recovery of the twitch response.
Total Duration of Action: In reference to the use of neuromuscular blocking agents, the time from drug administration to 90% recovery of the train-of-four twitch response.
Train-of-Four Ratio: A term used in reference to neuromuscular blocking agents in which a comparison is made between the fourth twitch of the train-of-four with the first twitch; when the fourth twitch is 90% of the first twitch, recovery from the neuromuscular blocking agent is indicated.
Physiology of neuromuscular transmission
Because of the frequent and routine intraoperative and postoperative use of drugs that alter neuromuscular function, a review of the anatomy and physiology of the neuromuscular system is important, with an emphasis on the chemical changes that occur at the receptor sites. Activation of skeletal muscle is both an electric and a biochemical event. The term conduction refers to the passage of an impulse along an axon to a muscle fiber. Transmission applies to passage of a neurotransmitter substance across a synaptic cleft (neuromuscular junction). The combined electric and chemical event is called neurohumoral transmission.1
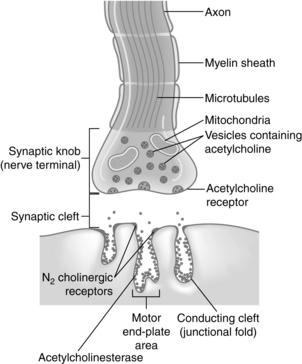
As the fine terminal branch of a motor neuron approaches the muscle fiber, it loses its myelin sheath and forms an expanded terminal that lies close to a specialized area of muscle membrane called the end plate (Fig. 23-1). Between the end of the muscle fiber and the end plate is the synaptic cleft, or neuromuscular junction. This space between the nerve and muscle fibers is approximately 20 nm wide. Acetylcholine is the biochemical neurotransmitter involved in the initiation of muscle contraction. Acetylcholine or cholinergic receptors are classified as nicotinic and muscarinic, respectively. The acetylcholine receptors are stimulated by acetylcholine. Anticholinesterase drugs such as neostigmine (Prostigmin), edrophonium chloride (Tensilon, Enlon), and pyridostigmine (Regonol) produce an increase in acetylcholine at the acetylcholine receptor. Therefore the pharmacologic effects of the anticholinesterase drugs are on both the nicotinic and muscarinic receptors. The nicotinic receptors are further classified as either N1 or N2 receptors. The N1 receptors are located at the presynaptic cleft and influence the release of acetylcholine. The N2 receptors are situated on the postsynaptic cleft in the neuromuscular junction and, when occupied by acetylcholine, open their channels to allow the flow of ions down the cell membrane, thus resulting in the skeletal muscle contraction. The nondepolarizing neuromuscular blocking agents such as pancuronium (Pavulon) produce a block of the N2 receptor and thus cause an inability of the channel to conduct ions, which results in skeletal muscle paralysis. Extrajunctional nicotinic receptors are located throughout the skeletal muscles. Their activity is normally suppressed by normal neural activity. However, when a patient has prolonged sepsis, inactivity, denervation, or burn trauma in the skeletal muscles, a proliferation of these extrajunctional nicotinic receptors results. As a result, these patients usually have an exaggerated hyperkalemic response when succinylcholine is administered.1
The muscarinic receptors are also subdivided into M1 and M2 receptors. M1 receptors are located in the autonomic ganglia and the central nervous system, and M2 receptors are located in the heart and salivary glands. Atropine and glycopyrrolate (Robinul) block both the M1 and the M2 receptors.2
Acetylcholine is formed in the body of the nerve cell and the cytoplasm of the nerve terminal and is stored in the small membrane-enclosed vesicles for subsequent release. A quantum is the amount of acetylcholine stored in each vesicle and represents approximately 10,000 molecules of acetylcholine. The presynaptic membrane contains discrete areas of specialization that are thought to be sites of release of the transmitter. These presynaptic active zones lie directly opposite the N2 cholinergic receptors, which are located on the postsynaptic membrane. This alignment ensures that the acetylcholine diffuses directly to the N2 receptors on the postsynaptic membrane quickly and in a high concentration. The N2 receptor, which responds to the neurotransmitter acetylcholine, is a glycoprotein that is an integral part of the postsynaptic membrane of the neuromuscular junction (see Fig. 23-1). New evidence indicates that a positive feedback mechanism also exists at the neuromuscular junction. Acetylcholine has a presynaptic action; therefore acetylcholine receptors are located on the presynaptic membrane. This positive feedback mechanism enhances the mobilization and release of acetylcholine. Finally, the enzyme that hydrolyzes acetylcholine is acetylcholinesterase, which is located in the neuromuscular junction.3
The initiation of skeletal muscle contraction occurs as a result of applying a threshold stimulus. An action potential that travels down the axon causes depolarization of the presynaptic membrane. As a result of this depolarization, the membrane permeability for calcium ions is increased and the calcium enters, or influxes, into the presynaptic membrane. Calcium acts to unite the vesicle to the presynaptic membrane and causes the rupture of that coalesced membrane, thus releasing acetylcholine into the fluid of the synaptic cleft (Fig. 23-2).
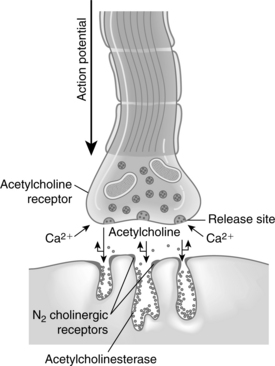
The acetylcholine molecules released from the nerve terminal into the synaptic cleft are subject to two main processes: (1) attachment to N2 cholinergic receptors located on the postsynaptic membrane, which leads to an opening of calcium channels that results in the movement of sodium into the region and generates an end plate potential (EPP); and (2) attachment of acetylcholine to the presynaptic nicotinic receptor, which enhances the release of more acetylcholine. When enough EPPs are generated, an action potential is propagated and spreads throughout the muscle and causes a change in the ionic permeability of the muscle sarcolemma. This process results in the release of calcium from the sarcoplasmic reticulum with a resultant increase in free calcium concentration in the muscle fiber. The process of excitation-contraction (E-C) coupling then takes place within that skeletal muscle cell. The physiologic outcome of E-C coupling is the contraction of the skeletal muscle. The increased concentration of calcium in the muscle fiber leads to an interaction between troponin-tropomyosin and actin. This interaction causes the active sites on actin to be exposed and interact with myosin and slide together, thus resulting in muscle contraction. This sliding of actin and myosin is sometimes called the ratchet effect.3 The contraction of the muscle fibers is terminated when calcium is pumped back into the sarcoplasmic reticulum of the muscle fibers. The calcium is stored in the sarcoplasmic reticulum for use when another action potential is generated.
Regulation and control of skeletal muscle contraction are also based on the enzymatic breakdown of acetylcholine. As previously discussed, the stimulus must be strong enough to release enough acetylcholine to bind to the postsynaptic N2 cholinergic receptor. This process of competition between the postsynaptic N2 receptor and acetylcholinesterase allows for some degree of regulation of the excitation process and for the recovery of the muscle cell membrane. The molecules of acetylcholine either diffuse in a random fashion to the N2 receptor or are destroyed by acetylcholinesterase. As the concentration gradient begins to decrease because of the destruction of acetylcholine by acetylcholinesterase, the N2 receptor gives up its acetylcholine, which is then destroyed, and the skeletal muscle relaxes. A small portion of the acetylcholine can escape the acetylcholinesterase in the synaptic cleft and migrate into the extracellular fluid and from there into the plasma. Acetylcholine within the plasma is then destroyed by plasma acetylcholinesterase, or pseudocholinesterase, which is produced in the liver.1
Pharmacologic overview of the skeletal muscle relaxants
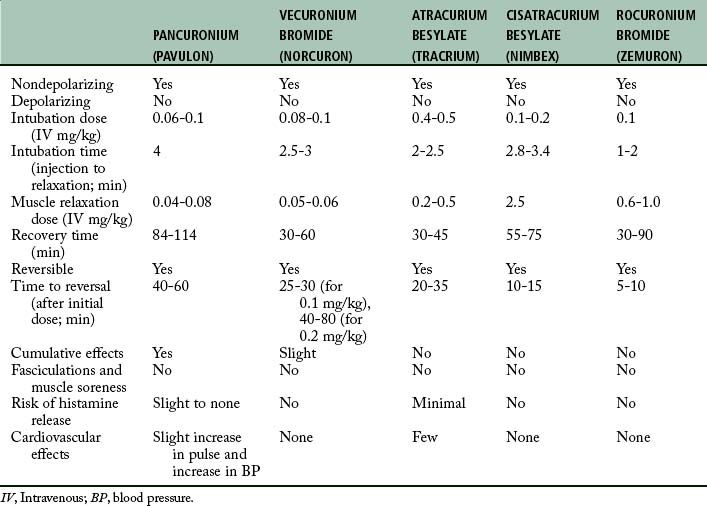
With the anatomy and physiology of neuromuscular transmission as background, the principal pharmacologic actions of the nondepolarizing and depolarizing skeletal muscle relaxants are discussed. Table 23-1 presents a pharmacologic overview of the commonly used skeletal muscle relaxants.
The prototypical nondepolarizing skeletal muscle relaxants are pancuronium and vecuronium (Norcuron). Pancuronium is an inhibitor of acetylcholine, is chemically viewed as two acetylcholine-like fragments, and has a bulky inflexible nucleus. This drug attaches to the N2 cholinergic receptors on the postsynaptic membrane and prevents depolarization. The skeletal muscle relaxant vecuronium has a chemical structure that is similar to a monoquaternary compound. The principal pharmacologic action of this drug is to block the postsynaptic N2 cholinergic receptor; in this way, it stops acetylcholine from binding to the receptor, which results in a competitive neuromuscular blockade. The nondepolarizing skeletal muscle relaxants also block the presynaptic cholinergic receptor and thus result in binding of the acetylcholine and thereby prevent activation of the positive feedback mechanism.4
The principal depolarizing skeletal muscle relaxant is succinylcholine (Anectine, Sucostrin). The molecular structure of this drug resembles two acetylcholine molecules back to back. Because of this structure, succinylcholine has the same effects as acetylcholine. Like acetylcholine, the succinylcholine molecule has a quaternary ammonium portion that is positively charged. This positively charged molecule is attracted by electrostatic action to the negatively charged N2 receptor. When the succinylcholine attaches to the receptor, a brief period of depolarization occurs that is manifested by transient muscular fasciculations. Succinylcholine also attaches to and activates the presynaptic acetylcholine receptor. This activation has an immediate effect of increased mobilization of acetylcholine in the motor nerve terminals, which explains why fasciculations are commonly observed after the administration of an intravenous bolus of succinylcholine. After the depolarization of the N2 receptor takes place, succinylcholine promotes and maintains the receptor in a depolarized state and prevents repolarization. Succinylcholine has a brief duration of action because of its rapid hydrolysis of the succinylcholine by the enzyme pseudocholinesterase, which is contained in the liver and plasma. The actions of succinylcholine cannot be pharmacologically reversed.5
Nondepolarizing neuromuscular blocking agents
Long-acting nondepolarizing skeletal muscle relaxants
Pancuronium bromide
Pancuronium bromide does not produce ganglionic blockade, but it does block the M2 cholinergic receptors in the heart. Consequently, when pancuronium bromide is administered, a slight 10% to 15% increase in heart rate is observed.6 Pancuronium activates the sympathetic nervous system by promoting the release of norepinephrine and blocking its uptake at the adrenergic nerve endings. After administration of this drug, a modest increase in mean arterial pressure and cardiac output is produced. Although isolated cases of histamine release have been reported, pancuronium can probably be used in patients who have a marginal allergy history. Pancuronium bromide is compatible with anesthetic agents used clinically and is safe for use in most patients when a nondepolarizing skeletal muscle relaxant is indicated. However, pancuronium bromide is not indicated when a nondepolarizing muscle relaxant is to be used with caution. In addition, pancuronium should not be used in patients who are undergoing chronic digitalis therapy because cardiac dysrhythmias have been reported. Finally, myocardial ischemia has been reported in patients with coronary artery disease when pancuronium is used. This ischemia is probably associated with the cardiac acceleration properties of the drug.
Intermediate-acting nondepolarizing skeletal muscle relaxants
Vecuronium bromide
Vecuronium (Norcuron) is a nondepolarizing skeletal muscle relaxant with a more rapid onset of action and a shorter duration of action than pancuronium. Actually, vecuronium is pancuronium without the quaternary methyl group in the steroid nucleus. Because of this structural difference, vecuronium has no effect on heart rate, arterial pressure, autonomic ganglia, or the alpha and beta adrenal receptors. The potency of vecuronium is equal to or slightly greater than that of pancuronium. Vecuronium has little or no cumulative effect. Although a portion of vecuronium is metabolized, most of the drug is excreted unchanged in the urine and bile. However, the neuromuscular blockade produced by vecuronium is not prolonged by renal failure. The duration of neuromuscular blockade produced by vecuronium is increased in patients with impaired hepatic function. Of clinical interest is that vecuronium, like atracurium, is less influenced by general inhalation anesthetics than is pancuronium.7 The pharmacologic action of this drug is easily reversed with the combination of an anticholinesterase and an anticholinergic drug.
The onset of action of vecuronium is between 2.5 and 3 minutes, with the normal dose of 0.08 mg/kg intravenously. Because of the rapid onset of action, vecuronium can be used for rapid-sequence intubation. In this instance, a doubling of the dose of vecuronium to 0.2 mg/kg can be used to achieve intubation conditions within 45 seconds to 2 minutes. Another method for use of vecuronium for intubation is the priming technique. The object of this technique is to administer a small priming dose of vecuronium several minutes before the intubation dose is given to shorten the onset of neuromuscular blockade. The usual priming dose is 0.015 mg/kg; after 3 minutes, an intubation dose of 0.1 mg/kg is administered. The onset of neuromuscular blockade should be between 70 and 90 seconds. The main drawback of this technique is the potential development of symptoms of partial neuromuscular blockade. Sensations reported are heavy eyelids, blurred vision, and difficulty in swallowing. Therefore, if this technique is used in the PACU, the nurse should warn patients of the possible symptoms, and ventilatory support should always be available.
Because of concerns about the priming technique, the timing technique was developed. In the timing technique, which is used mainly in the operating room, the patient is given vecuronium before sodium pentothal. Consequently, the induction of anesthesia is specifically timed to the onset of clinical muscular weakness. In this technique, the patient is given 0.1 to 0.2 mg/kg of vecuronium intravenously. At the onset of weakness, as determined with the peripheral nerve simulator, a 1.0- to 2.5-mg/kg bolus dose of propofol is given. Intubating conditions occur within 1 minute.4
Atracurium besylate
Atracurium is less potent than pancuronium and has a rapid onset of 1 to 3 minutes and a duration of action of 30 to 45 minutes. For endotracheal intubation in the PACU setting, 0.3 to 0.5 mg/kg of atracurium should provide adequate skeletal muscle relaxation for intubation in about 2.5 minutes. For maintenance of mechanical ventilation in the PACU setting, an infusion rate of 10 mcg/kg/min of atracurium may be used. When the infusion has been discontinued, spontaneous ventilation by the patient occurs in approximately 30 minutes.1 The effects can be reversed with a combination of anticholinesterase and antimuscarinic in 12 to 15 minutes after the discontinuation of the atracurium infusion.
Cisatracurium besylate
The average adult intubation dose of cisatracurium is 0.2 mg/kg and has an onset of approximately 90 seconds, a peak in 3 to 5 minutes, and a duration of action of 40 to 50 minutes. A supplemental dose of 0.03 mg/kg provides an additional 20 minutes of skeletal muscle relaxation. For maintenance of a stable state of skeletal muscle relaxation in the PACU, cisatracurium can be administered via infusion at a rate of 1 to 2 mcg/kg/min.8
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


