*Administration of a 250- to 500-mg supplemental dose following dialysis is recommended.
■ Information is unavailable for dosage adjustments in pediatric patients with renal impairment.
Dilution
Adults:
Each vial contains 500 mg/5 mL (100 mg/mL). A single dose (500, 1,000, or 1,500 mg [1, 2, or 3 vials]) must be diluted in 100 mL of NS, LR, or D5W for infusion. Doses of 500, 1,000, or 1,500 mg are available prediluted in 100 mL of NS.
Pediatric patients:
A smaller volume of infusion solution may be required due to limitations around the total daily fluid intake of the patient. The amount of diluent should be calculated such that the final concentration of the levetiracetam solution does not exceed 15 mg/mL. For example, a 1-year-old 10-kg child beginning therapy for treatment of partial-onset seizures would initiate therapy at 20 mg/kg/day = 200 mg/day = 100 mg twice daily. 100 mg (1 mL) mixed in 24 mL of compatible solution will provide a final concentration of 4 mg/mL. In a fluid-restricted patient, the solution volume could be decreased to as low as 5.7 mL [100 mg ÷ (1 mL from drug + 5.7 mL of solution) = 14.9 mg/mL].
Filters:
Specific information not available.
Storage:
Store at CRT. Diluted solution stable for at least 24 hours at CRT stored in polyvinyl chloride (PVC) bags. Discard any unused portion of vial.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer lists solutions NS, LR, and D5W and other antiepileptic drugs lorazepam (Ativan), diazepam (Valium), and valproate sodium (Depacon) as compatible for at least 24 hours stored in polyvinyl chloride (PVC) bags at CRT.
Rate of administration
A single dose properly diluted as an infusion over 15 minutes.
Actions
An antiepileptic (anticonvulsant) drug. The precise mechanism of action is unknown. In animal studies it inhibited burst firing without affecting normal neuronal excitability, which suggests that it may selectively prevent hypersynchronization of epileptiform burst firing and propagation of seizure activity in human complex partial seizures. Less than 10% bound to plasma proteins. Not extensively metabolized in humans and not liver (cytochrome P450) dependent. Undergoes enzymatic hydrolysis. Half-life is 6 to 8 hours. Primarily excreted in urine as unchanged drug and metabolites. Crossed the placental barrier in animal studies. Secreted in breast milk.
Indications and uses
IV injection is used as a temporary alternative for patients when oral administration is temporarily not feasible. ■ Adjunctive therapy in the treatment of partial-onset seizures in adults and pediatric patients 1 month of age and older with epilepsy. ■ Adjunctive therapy in the treatment of myoclonic seizures in adults and adolescents 12 years of age and older with juvenile myoclonic epilepsy (JME). ■ Adjunctive therapy in the treatment of primary generalized tonic-clonic seizures in adults and pediatric patients 6 years of age and older with idiopathic generalized epilepsy.
Contraindications
Manufacturer states, “None.”
Precautions
For IV use only. ■ Associated with CNS side effects (somnolence and fatigue, coordination difficulties [abnormal gait, ataxia, incoordination], and behavioral abnormalities [e.g., aggression, agitation, anxiety, apathy, depression, emotional lability, irritability, and psychotic symptoms, including psychosis and suicidal tendencies]). Somnolence and fatigue and coordination difficulties occurred most frequently within the first 4 weeks of treatment and resolved or improved with dose reduction or discontinuation of levetiracetam. ■ Antiepileptic drugs (AEDs) increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior. Some behavioral changes resolved without intervention. Others required dose reduction or discontinuation of levetiracetam. ■ Serious dermatologic reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis, have been reported. Median time to onset is 14 to 17 days, but development up to 4 months after initiation of therapy has been reported. ■ To minimize the potential of increased seizure frequency, withdraw antiepileptic drugs (including levetiracetam) gradually. ■ Increase in diastolic BP has been reported in patients 1 month to under 4 years of age who are receiving the oral formulation of levetiracetam. This side effect was not observed in older children or adults.
Monitor:
Baseline CBC and CrCl indicated; monitor as needed. Decreases in WBC, RBC, hemoglobin, and hematocrit have been reported, but a change or discontinuation of therapy was not required. Increases in eosinophil counts and cases of agranulocytosis have also been reported. ■ Monitor for seizure activity. ■ Observe patient closely for signs of CNS side effects; see Precautions. ■ Monitor vital signs. Pediatric patients 1 month to under 4 years of age should be monitored for a possible increase in diastolic BP. ■ Monitor for serious dermatologic reactions.
Patient education:
May cause dizziness, coordination difficulties, and somnolence; use caution when performing tasks that require alertness or coordination (e.g., operating machinery, driving). ■ Inform your health care professional if you are pregnant or breast-feeding. ■ May increase the risk of suicidal thoughts and behavior. Promptly report emergence or worsening of S/S of depression, any unusual changes in mood or behavior, or thoughts about self-harm. ■ Women who are pregnant or who become pregnant should be encouraged to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. ■ May cause serious dermatologic reactions; promptly report development of a rash.
Maternal/child:
Category C: animal studies demonstrated fetal skeletal abnormalities and delayed offspring growth. Use during pregnancy only if potential benefit justifies the potential risk to the fetus. ■ Physiologic changes during pregnancy may decrease levetiracetam concentration. Monitor carefully during pregnancy. Extend monitoring to postpartum period if dose adjustments were required during the pregnancy. ■ Effects during labor and delivery unknown. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in certain pediatric populations have been established; see Indications. ■ Pharmacokinetic analysis showed that body weight was significantly correlated to the clearance of levetiracetam in pediatric patients; clearance increased with an increase in body weight.
Elderly:
Safety similar to that seen in younger adults; however, total body clearance is decreased and half-life is increased. ■ Reduced doses may be indicated; see Dose Adjustments. Monitoring of renal function suggested.
Drug/lab interactions
Manufacturer states that levetiracetam “is unlikely to produce, or be subject to, pharmacokinetic interactions.” ■ Other antiepileptic drugs (AEDs) such as carbamazepine (Tegretol), gabapentin (Neurontin), lamotrigine (Lamictal), phenobarbital (Luminal), phenytoin (Dilantin), primidone (Mysoline), and valproate (Depacon) do not influence the pharmacokinetics of levetiracetam, and it does not influence their pharmacokinetics. ■ An increase in the clearance of levetiracetam was seen in pediatric patients when it was coadministered with enzyme-inducing AEDs (e.g., carbamazepine [Tegretol], phenytoin [Dilantin]). Levetiracetam had no effect on plasma concentrations of carbamazepine (Tegretol), lamotrigine (Lamictal), topiramate (Topamax), or valproate (Depacon) in pediatric patients. ■ Not an inhibitor or a substrate of cytochrome P450 isoforms; unlikely to have interactions with drugs metabolized by these isoenzymes in the liver. ■ Minimal protein binding; unlikely to have interactions with drugs requiring protein-binding sites. ■ Has been administered with oral contraceptives, digoxin (Lanoxin), and warfarin (Coumadin) with no interference on the pharmacokinetics of either drug. ■ Probenecid decreases renal clearance and increases serum concentrations of one of the metabolites of levetiracetam.
Side effects
Adults:
Asthenia, dizziness, infection, and somnolence occurred most frequently during the first 4 weeks of treatment.
Pediatric patients:
Aggression, decreased appetite, fatigue, irritability, and nasal congestion were most commonly reported.
Patients of all ages:
Other side effects reported and presenting anywhere from 1 to 4 weeks included amnesia; anorexia; anxiety; ataxia; cough; decreased RBC, WBC, Hct, and Hgb; depersonalization; depression; diarrhea; diplopia; emotional lability; headache; hostility; influenza; insomnia; irritability; mood swings; neck pain; nervousness; pain; paresthesia; pharyngitis; rhinitis; seizures; sinusitis; and vertigo.
Post-marketing:
Abnormal liver function tests, agranulocytosis, choreoathetosis, drug rash with eosinophilia and systemic symptoms (DRESS), dyskinesia, erythema multiforme, hepatic failure, hepatitis, hyponatremia, leukopenia, muscle weakness, neutropenia, pancreatitis, pancytopenia (with bone marrow suppression), panic attack, Stevens-Johnson syndrome, suicidal behavior, thrombocytopenia, toxic epidermal necrolysis, and weight loss. Alopecia has been reported; recovery occurred in the majority of cases if levetiracetam was discontinued.
Overdose:
Aggression, agitation, coma, depressed level of consciousness, drowsiness, respiratory depression, and somnolence.
Antidote
Keep physician informed of all side effects. Some may not require intervention, and others may improve with a reduced dose or discontinuation of levetiracetam; see Precautions and Side Effects. Discontinue at first sign of rash, unless the rash is clearly not drug related. Support patient as required in treatment of overdose. No specific antidote in overdose; however, hemodialysis will remove approximately 50% of a dose in 4 hours.
Levofloxacin 
(lee-voh-FLOX-ah-sin)
Levaquin
Antibacterial (fluoroquinolone)
pH 3.8 to 5.8
Usual dose
250 to 750 mg once every 24 hours.* Dose and duration of treatment are based on degree of infection and specific diagnosis. CrCl equal to or greater than 50 mL/min is required. Dose and serum levels similar by oral or IV route. Transfer to oral dose as soon as practical.
| Levofloxacin Dosing Guidelines | ||
| Type of Infection a | Dose Every 24 Hours b | Duration (Days) |
| Acute Bacterial Exacerbation of Chronic Bronchitis | 500 mg | 7 days |
| Nosocomial Pneumonia | 750 mg | 7-14 days |
| Community-Acquired Pneumonia | 500 mg or 750 mg | 7-14 days 5 days |
| Acute Bacterial Sinusitis | 500 mg or 750 mg | 10-14 days 5 days |
| Chronic Bacterial Prostatitis | 500 mg | 28 days |
| Complicated SSSI c | 750 mg | 7-14 days |
| Uncomplicated SSSI c | 500 mg | 7-10 days |
| Complicated UTI d or Acute Pyelonephritis | 750 mg | 5 days |
| Complicated UTI d or Acute Pyelonephritis | 250 mg | 10 days |
| Uncomplicated UTI d | 250 mg | 3 days |
| Inhalation Anthrax (postexposure) and plague in adults e | 500 mg | 60 days (anthrax) 10 to 14 days (plague) |
| Inhalation Anthrax (postexposure) and plague in pediatric patients >50 kg e | 500 mg | 60 days (anthrax) 10 to 14 days (plague) f |
| Inhalation Anthrax (postexposure) and plague in pediatric patients <50 kg and ≥6 months of age e | 8 mg/kg q 12 hr b (not to exceed 250 mg/dose) | 60 days (anthrax) 10 to 14 days (plague) f |
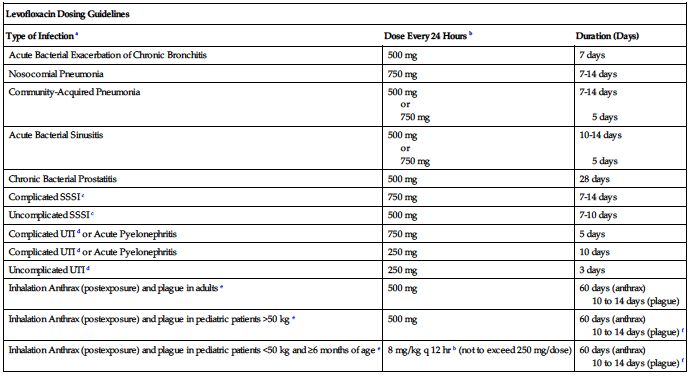
aDue to the designated pathogens (see Indications and Usage).
bFrequency is every 12 hours (not 24 hours) for treatment of inhalation anthrax and plague in pediatric patients less than 50 kg and age 6 months or more.
cSkin and skin structure infections.
eBegin drug administration as soon as possible after suspected or confirmed exposure to aerosolized Bacillus anthracis or to Yersinia pestis. If clinically indicated, higher doses of levofloxacin (e.g., equivalent to 750 mg) can be used for the treatment of plague.
Dose adjustments
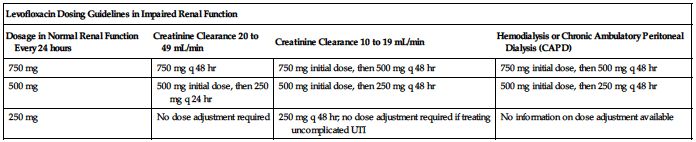
No dose adjustment is required specifically for age, gender, race, or in impaired hepatic function. ■ Clearance is reduced and half-life is prolonged in patients with a CrCl equal to or less than 50 mL/min. See the following chart for dosing guidelines. See Important IV Therapy Facts on p. xx for formula to convert SCr to CrCl. ■ Supplemental doses are not required after hemodialysis or peritoneal dialysis.
| Levofloxacin Dosing Guidelines in Impaired Renal Function | |||
| Dosage in Normal Renal Function Every 24 hours | Creatinine Clearance 20 to 49 mL/min | Creatinine Clearance 10 to 19 mL/min | Hemodialysis or Chronic Ambulatory Peritoneal Dialysis (CAPD) |
| 750 mg | 750 mg q 48 hr | 750 mg initial dose, then 500 mg q 48 hr | 750 mg initial dose, then 500 mg q 48 hr |
| 500 mg | 500 mg initial dose, then 250 mg q 24 hr | 500 mg initial dose, then 250 mg q 48 hr | 500 mg initial dose, then 250 mg q 48 hr |
| 250 mg | No dose adjustment required | 250 mg q 48 hr; no dose adjustment required if treating uncomplicated UTI | No information on dose adjustment available |
Dilution
Available in single-use vials and as prediluted, ready-to-use infusions.
Single-use vials:
Withdraw desired dose from single-use vial (10 mL for 250 mg, 20 mL for 500 mg, 30 mL for 750 mg). Each 10 mL (250 mg) must be further diluted with a minimum of 40 mL NS, D5W, D5NS, D5LR, D5/plasma-Lyte 56, D5/1/2NS with 0.15% KCl, or 1/6 M sodium lactate. Desired concentration is 5 mg/mL. No preservatives; enter vial only once. When 500-mg (20-mL) vial is used to prepare two 250-mg doses, withdraw entire contents of vial at once using a single-entry procedure. Prepare and store second dose for subsequent use.
Premix flexible containers:
No further dilution necessary. Available as 250 mg in 50 mL, 500 mg in 100 mL, or 750 mg in 150 mL D5W. Instructions for access to and use of premix flexible containers are on its storage carton. Do not use flexible containers in series connections.
Filters:
Not required; however, contents of both vials and premixed solutions were filtered during manufacturing with polyvinyl mixed ester cellulose filters. Size not specified by manufacturer. No significant loss of potency expected.
Storage:
Store vials at CRT; protect from light. Store premix at or below 25° C (77° F); protect from freezing, light, and excessive heat. Brief exposure up to 40° C (104° F) does not adversely affect the product. Both are stable to expiration date. Solutions diluted from vials are stable at or below 25° C (77° F) for 3 days and for up to 14 days if refrigerated. May be frozen for up to 6 months. Do not force thaw (e.g., microwave or water bath) and do not refreeze. Discard any unused portion of premixed solutions and/or opened vials.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Limited compatibility information available; other intravenous substances, additives, or other medications should not be added to levofloxacin or infused simultaneously through the same intravenous line.” Never administer in the same IV or through the same tubing with any solution containing multivalent cations (e.g., calcium, magnesium). Flush line with a compatible solution before and after administration of levofloxacin and/or any other drug through the same IV line.
One source suggests the following compatibilities:
Y-site:
Amikacin, aminophylline, ampicillin, anidulafungin (Eraxis), bivalirudin (Angiomax), caffeine citrate (Cafcit), caspofungin (Cancidas), cefotaxime (Claforan), ceftaroline (Teflaro), clindamycin (Cleocin), daptomycin (Cubicin), dexamethasone (Decadron), dexmedetomidine (Precedex), dobutamine, dopamine, doripenem (Doribax), epinephrine (Adrenalin), fenoldopam (Corlopam), fentanyl, gentamicin, hetastarch in electrolytes (Hextend), hydromorphone (Dilaudid), 6% hydroxyethyl starch (Voluven), insulin (regular), isoproterenol (Isuprel), lidocaine, linezolid (Zyvox), lorazepam (Ativan), magnesium sulfate, metoclopramide (Reglan), morphine, oxacillin (Bactocill), pancuronium, penicillin G sodium, phenobarbital (Luminal), phenylephrine (NeoSynephrine), potassium chloride, sodium bicarbonate, vancomycin.
Rate of administration
Each 250- or 500-mg dose must be equally distributed over 60 minutes as an infusion. A 750-mg dose must be equally distributed over 90 minutes as an infusion. Too-rapid administration may cause hypotension. May be given through a Y-tube or three-way stopcock of infusion set. Temporarily discontinue other solutions infusing at the same site and flush tubing with compatible solutions before and after levofloxacin.
Actions
A synthetic, broad-spectrum, fluoroquinolone antibacterial agent. Bactericidal to aerobic gram-negative and gram-positive organisms through interference with an enzyme (topoisomerase II) needed for synthesis of bacterial DNA. May be active against bacteria resistant to aminoglycosides, beta-lactam antibiotics, and macrolides. Onset of action is prompt, and serum levels are dose related. Mean terminal half-life is 6 to 8 hours. Steady state is achieved within 48 hours. Widely distributed into body tissues, including blister fluid and lung tissues. Moderately bound to serum protein (24% to 38%). Metabolism is minimal; primarily excreted as unchanged drug in urine. Very small amounts found in bile and feces. May cross placental barrier. May be secreted in breast milk.
Indications and uses
Treatment of adults with mild, moderate, and severe infections caused by susceptible strains of microorganisms in conditions including acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and nosocomial pneumonia. ■ Treatment of community-acquired pneumonia due to many organisms, including Streptococcus pneumoniae (including multidrug-resistant strains [MDRSP]). MDRSP strains are resistant to two or more of the following antibiotics: penicillin, second-generation cephalosporins (e.g., cefuroxime [Zinacef]), macrolides, tetracyclines, and sulfamethoxazole/trimethoprim. ■ Treatment of complicated skin and skin structure infections, mild to moderate uncomplicated skin and skin structure infections, complicated and uncomplicated urinary tract infections, and acute pyelonephritis (including cases with concurrent bacteremia). ■ To reduce the incidence and progression of inhalational anthrax following exposure to Bacillus anthracis. Begin administration as soon as possible after suspected or confirmed exposure. Transfer to oral therapy when practical. ■ Treatment of plague, including pneumonic and septicemic plague, due to Yersinia pestis, as well as prophylaxis for plague in adults and pediatric patients 6 months of age and older. ■ Treatment of chronic bacterial prostatitis (usually oral therapy).
Contraindications
History of hypersensitivity to levofloxacin, its components, or any other quinolone antimicrobial agents (e.g., ciprofloxacin [Cipro], norfloxacin [Noroxin]).
Precautions
For IV use only. ■ Culture and sensitivity studies indicated to determine susceptibility of the causative organism to levofloxacin. ■ Pseudomonas aeruginosa may develop resistance during treatment. Ongoing culture and sensitivity studies indicated. ■ The emergence of bacterial resistance to fluoroquinolones and the occurrence of crossresistance with other fluoroquinolones have been observed and are of concern. Proper use of fluoroquinolones and other classes of antibiotics is encouraged to avoid the emergence of resistant bacteria from overuse. ■ Prolonged use may cause superinfection because of overgrowth of nonsusceptible organisms. ■ Use with caution in patients with known CNS disorders that predispose to seizures or alter seizure threshold (e.g., epilepsy, severe cerebral arteriosclerosis, concomitant drug therapy). Convulsions, toxic psychosis, increased intracranial pressure (including pseudotumor cerebri), and CNS stimulation have been reported. May cause other CNS effects, including confusion, depression, dizziness, hallucinations, tremors and, rarely, suicidal thoughts or acts. ■ Use caution in patients with impaired renal function; see Dose Adjustments. ■ May be used by patients who are allergic to penicillin or intolerant of macrolides (e.g., erythromycin). ■ Cross-resistance may occur with other fluoroquinolones, but some microorganisms resistant to other fluoroquinolones may be susceptible to levofloxacin. ■ Severe, sometimes fatal hepatotoxicity has been reported. Symptoms appeared within 6 to 14 days of initiating therapy, and most cases were not associated with hypersensitivity and occurred in patients 65 years of age or older; see Patient Education and Antidote. ■ Tendinitis and tendon rupture that required surgical repair or resulted in prolonged disability have been reported in patients of all ages receiving quinolones. Most frequently involves the Achilles tendon but has also been reported with the shoulder, hand, biceps, thumb, and other tendon sites. ■ Tendon rupture or tendinitis may occur during or up to months after fluoroquinolone therapy. Risk may be increased in patients over 60 years of age; in patients taking corticosteroids; in patients with heart, kidney, or lung transplants; with strenuous physical activity; and in patients with renal failure or previous tendon disorders such as rheumatoid arthritis. ■ Prolongation of the QT interval on ECG and infrequent cases of arrhythmia (including torsades de pointes) have been reported. The risk of arrhythmia may be reduced by avoiding the use of levofloxacin in patients with known prolongation of the QT interval, uncorrected electrolyte imbalances (e.g., hypokalemia, hypomagnesemia), significant bradycardia, cardiomyopathy, or concurrent treatment with Class Ia antiarrhythmic agents (e.g., quinidine [Quinidex], procainamide [Procanbid, Procan SR]) or Class III antiarrhythmic agents (e.g., amiodarone [Nexterone], sotalol [Betapace]) and any other drug that can prolong the QT interval; avoid coadministration; see Drug/Lab Interactions. ■ Fluoroquinolones have neuromuscular blocking activity and may exacerbate muscle weakness in persons with myasthenia gravis. Serious adverse events, including a requirement for ventilatory support and deaths, have been reported in these patients. Avoid use in patients with a known history of myasthenia gravis. ■ Rare cases of peripheral neuropathy (e.g., paresthesias, hypoesthesias, dysesthesias [impairment of sensitivity or touch], or weakness) have been reported. Symptoms may occur soon after initiation of therapy and may be irreversible; see Antidote. ■ Clostridium difficile–associated diarrhea (CDAD) has been reported. May range from mild diarrhea to fatal colitis. Consider in patients who present with diarrhea during or after treatment with levofloxacin. ■ Serious and occasionally fatal hypersensitivity and/or anaphylactic reactions have been reported. Often occur after the first dose. ■ Other serious events (sometimes fatal) due to hypersensitivity or uncertain etiology have been reported with fluoroquinolones, including levofloxacin; usually occur after multiple doses. Manifestations may include renal impairment/failure, hematologic toxicity, and dermatologic toxicity; see Side Effects, Post-Marketing. Discontinue levofloxacin at the first appearance of a skin rash, jaundice, or other signs of hepatotoxicity or hypersensitivity. ■ Moderate to severe photosensitivity/phototoxicity reactions have been reported in patients receiving quinolones; see Patient Education. ■ Not tested in humans for postexposure prevention of inhalation anthrax; however, plasma concentrations are considered to be in a range to produce effective results. ■ Safety for use in adults beyond 28 days or in pediatric patients beyond 14 days not studied; use only for prescribed indication; benefits must outweigh risks. An increased incidence of musculoskeletal adverse events has been observed in pediatric patients.
Monitor:
Hypersensitivity reactions, including anaphylaxis with the first or succeeding doses, have been reported in patients receiving quinolones, even in those without known hypersensitivity. Emergency equipment must always be available; see Precautions. ■ Obtain baseline CBC with differential, CrCl, and blood glucose. Periodic monitoring of organ systems, including hematopoietic, hepatic, and renal, is recommended. ■ Monitor for S/S of peripheral neuropathy. Discontinue levofloxacin at the first symptoms of neuropathy (e.g., pain, burning, tingling, numbness and/or weakness) or if patient is found to have deficits in light touch, pain, temperature, position sense, vibratory sensation, and/or motor strength. ■ Maintain adequate hydration to prevent concentrated urine throughout treatment. Crystalluria and cylindruria have been reported with quinolones. ■ Monitor infusion site for inflammation and/or extravasation. ■ Symptomatic hyperglycemia or hypoglycemia may occur, usually in diabetic patients receiving oral hypoglycemic agents (e.g., glyburide) or insulin. Monitor blood glucose closely. ■ See Precautions, Drug/Lab Interactions, and Antidote.
Patient education:
A patient medication guide is available from the manufacturer. ■ Review all medicines and disease history with pharmacist or physician before initiating treatment. ■ Inform physician of any history of myasthenia gravis. ■ Patients with a history of myasthenia gravis should avoid the use of levofloxacin. ■ Drink fluids liberally. ■ Promptly report skin rash or any other hypersensitivity reaction. ■ Promptly report pain, burning, tingling, numbness and/or weakness in extremities. Nerve damage can be permanent. ■ Photosensitivity has occurred in a minimal number of patients, but it is best to avoid excessive sunlight or artificial ultraviolet light. May cause severe sunburn; wear protective clothing, use sunscreen, and wear dark glasses outdoors. Report a sunburn-like reaction or skin eruption promptly. ■ Dizziness or light-headedness may interfere with ambulation and motor coordination. Use caution in tasks that require alertness. ■ Effects of caffeine, theophylline preparations, and/or warfarin (Coumadin) may be increased; notify your physician if you take any of these agents. If diabetic and on medication, monitor your blood glucose carefully. If a hypoglycemia reaction occurs, discontinue levofloxacin and consult physician. ■ Promptly report S/S of liver injury (e.g., dark-colored urine, fever, itching, jaundice [yellowing of skin or whites of eyes], light-colored bowel movements, loss of appetite, nausea, right upper quadrant tenderness, tiredness, vomiting, weakness). ■ Promptly report tendon pain or inflammation; rest and refrain from exercise. ■ Before initiating therapy, parents should inform their child’s physician if their child has a history of joint-related problems, and they should notify their child’s physician of any tendon or joint-related problems that occur during or after therapy. ■ Promptly report diarrhea or bloody stools that occur during treatment or up to several months after an antibiotic has been discontinued; may indicate CDAD and require treatment. ■ See Precautions, Monitor, Drug/Lab Interactions, and Antidote.
Maternal/child:
Category C: safety for use in pregnancy not established; benefits must outweigh risks. ■ Discontinue breast-feeding. ■ Safety for use in pediatric patients under 18 years of age not established. Indicated in pediatric patients 6 months of age or older only for prevention of inhalation anthrax (postexposure) and prevention and treatment of plague. Safety for use in pediatric patients under 6 months of age not established. An increased incidence of musculoskeletal disorders (arthralgia, arthritis, tendinopathy, and gait abnormality) compared with controls has been observed in pediatric patients receiving levofloxacin.
Elderly:
Half-life may be slightly extended due to age-related renal impairment. Dose reduction required only in the elderly with a CrCl of 50 mL/min or less. ■ Safety and effectiveness similar to that in younger adults; however, they may experience an increased risk of side effects (e.g., CNS effects, hepatotoxicity, tendinitis, tendon rupture, risk of QT prolongation). Monitoring of renal function may be useful. ■ See Dose Adjustments and Precautions.
Drug/lab interactions
May cause ventricular arrhythmias or torsades de pointes with drugs that prolong the QT interval, such as Class Ia antiarrhythmic agents (e.g., quinidine, procainamide [Procanbid, Procan SR]), Class III antiarrhythmic agents (e.g., amiodarone [Nexterone], sotalol [Betapace]), anticonvulsants (e.g., fosphenytoin [Cerebyx]), antihistamines (e.g., diphenhydramine [Benedryl]), phenothiazines (e.g., chlorpromazine [Thorazine]), serotonin reuptake inhibitors (e.g., fluoxetine [Prozac], paroxetine [Paxil]), tricyclic antidepressants (e.g., imipramine [Tofranil], amitryptyline [Elavil]). Avoid coadministration. ■ Risk of CNS stimulation and convulsive seizures may be increased with NSAIDs (e.g., ibuprofen [Advil, Motrin], naproxen [Aleve, Naprosyn]). ■ May cause hyperglycemia and hypoglycemia with concurrent administration of antidiabetic agents (e.g., metformin [Glucophage], insulin); monitoring of blood glucose recommended. ■ Concomitant administration with cimetidine or probenecid reduces levofloxacin renal clearance by 24% and 35% respectively, but dose adjustment is not required. ■ Interactions with theophylline that occur with other quinolones have not been noted, but monitoring of theophylline levels is recommended with concomitant use. ■ Some quinolones, including levofloxacin, may enhance effects of warfarin (Coumadin); monitoring of PT or INR is recommended with concomitant use. ■ No dose adjustment required for either drug when levofloxacin is administered concomitantly with cyclosporine or digoxin. ■ See Precautions. ■ May cause false-positive when testing urine for opiates; more specific testing methods may be indicated.
Side effects
The most common side effects are constipation, diarrhea, dizziness, headache, insomnia, and nausea. Abdominal pain, chest pain, crystalluria, cylindruria, dyspepsia, dyspnea, edema, injection site reaction, moniliasis, photosensitivity/phototoxicity (sun sensitivity), pruritus, rash, vaginitis, and vomiting have occurred. Abnormal dreaming, abnormal gait, abnormal hepatic function, abnormal renal function, acute renal failure, agitation, anemia, anorexia, anxiety, arthralgia, cardiac arrest, CDAD, confusion, convulsions, depression, epistaxis, esophagitis, gastritis, gastroenteritis, genital moniliasis, glossitis, granulocytopenia, hallucinations, hyperglycemia, hyperkalemia, hyperkinesia, hypersensitivity reactions, hypertonia, hypoglycemia, increased alkaline phosphatase, increased hepatic enzymes, myalgia, nightmares, palpitation, pancreatitis, paresthesia, phlebitis, pseudomembranous colitis, skeletal pain, sleep disorders, somnolence, stomatitis, syncope, tendinitis, thrombocytopenia, tremor, urticaria, ventricular arrhythmia, ventricular tachycardia, and vertigo have also been reported in fewer than 1% of patients.
Post-marketing:
Abnormal EEG; hematologic abnormalities (agranulocytosis, anemia [hemolytic and aplastic], eosinophilia, leukopenia, pancytopenia, thrombocytopenia [including thrombotic thrombocytopenic purpura]); exacerbation of myasthenia gravis; eye disorders (e.g., blurred vision, diplopia, reduced visual acuity, scotoma, uveitis); hepatic failure, including fatal cases; fever; hepatitis; hypersensitivity reactions (sometimes fatal, including angioneurotic edema, anaphylaxis); interstitial nephritis; jaundice; leukocytoclastic vasculitis; multiorgan failure; muscle injury and increased muscle enzymes; paranoia; peripheral neuropathy; photosensitivity/phototoxicity reactions; prolonged INR; prolonged PT; prolonged QT interval; pseudotumor cerebri; psychosis; rhabdomyolysis; serum sickness; severe dermatologic reactions (e.g., toxic epidermal necrolysis [Lyell’s syndrome], Stevens-Johnson syndrome); tachycardia; tendon rupture; tinnitus; vasodilation; and isolated reports of allergic pneumonitis, encephalopathy, suicide attempts and suicidal ideation, and torsades de pointes.
Antidote
Keep physician informed of all side effects. Most minor side effects will be treated symptomatically; monitor closely. Discontinue levofloxacin at the first sign of any major side effect (CDAD, CNS symptoms, dermatologic reactions, hepatotoxicity, hypersensitivity, hypoglycemic reactions, phototoxicity, symptoms of neuropathy, or tendon rupture). Treat hypersensitivity reactions as indicated with epinephrine, airway management, oxygen, IV fluids, antihistamines (e.g., diphenhydramine [Benadryl]), corticosteroids (e.g., Solu-Cortef), and pressor amines (e.g., dopamine). Treat CNS symptoms as indicated; may require diazepam (Valium) for seizures. Mild cases of CDAD may respond to discontinuation of levofloxacin. Treat CDAD with fluids, electrolytes, protein supplements, and oral vancomycin (Vancocin) or metronidazole (Flagyl) as indicated. In severe cases, surgical evaluation may be indicated. Complete rest is indicated for an affected tendon until treatment is available. Maintain hydration in overdose. No specific antidote; not removed by hemodialysis or peritoneal dialysis. Maintain patient until drug is excreted and symptoms subside.
Levoleucovorin
(lee-voh-loo-koh-VOR-in)
Fusilev
Folate analog
Antidote
Antineoplastic adjunct
Usual dose
Dosed at one-half the usual dose of the racemic form (leucovorin calcium).
Levoleucovorin rescue after high-dose methotrexate therapy:
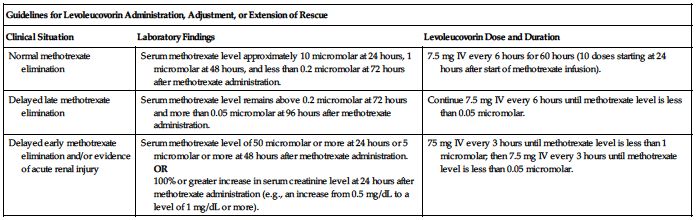
Dose is based on a methotrexate dose of 12 Gm/M2 infused over 4 hours; see methotrexate monograph. Obtain serum creatinine and methotrexate levels at least once daily. Additional hydration and urinary alkalinization (pH 7 or greater) is indicated and should be continued until the methotrexate level is less than 0.05 micromolar. Administer and/or adjust the levoleucovorin dose or extend rescue based on the following guidelines. See Dose Adjustments, Precautions, and Monitor.
| Guidelines for Levoleucovorin Administration, Adjustment, or Extension of Rescue | ||
| Clinical Situation | Laboratory Findings | Levoleucovorin Dose and Duration |
| Normal methotrexate elimination | Serum methotrexate level approximately 10 micromolar at 24 hours, 1 micromolar at 48 hours, and less than 0.2 micromolar at 72 hours after methotrexate administration. | 7.5 mg IV every 6 hours for 60 hours (10 doses starting at 24 hours after start of methotrexate infusion). |
| Delayed late methotrexate elimination | Serum methotrexate level remains above 0.2 micromolar at 72 hours and more than 0.05 micromolar at 96 hours after methotrexate administration. | Continue 7.5 mg IV every 6 hours until methotrexate level is less than 0.05 micromolar. |
| Delayed early methotrexate elimination and/or evidence of acute renal injury | Serum methotrexate level of 50 micromolar or more at 24 hours or 5 micromolar or more at 48 hours after methotrexate administration. OR 100% or greater increase in serum creatinine level at 24 hours after methotrexate administration (e.g., an increase from 0.5 mg/dL to a level of 1 mg/dL or more). | 75 mg IV every 3 hours until methotrexate level is less than 1 micromolar; then 7.5 mg IV every 3 hours until methotrexate level is less than 0.05 micromolar. |
Inadvertent methotrexate overdose:
7.5 mg IV every 6 hours (approximately 5 mg/M2) until the serum methotrexate level is less than 5 × 10−8 M (0.05 micromolar). Begin levoleucovorin rescue as soon as possible after an inadvertent methotrexate overdose and within 24 hours of methotrexate administration when there is delayed excretion. The effectiveness of levoleucovorin in counteracting toxicity may decrease as the time interval between methotrexate administration and levoleucovorin rescue increases. See Dose Adjustments and Monitor.
Colorectal cancer:
Several regimens are in use. See fluorouracil (5-FU) monograph for 5-FU specific information. Administer 5-FU and levoleucovorin separately to avoid the formation of a precipitate.
Flush the IV line with NS between drugs.
Levoleucovorin 100 mg/M2 by IV injection over a minimum of 3 minutes (see Rate of Administration) followed by 5-FU 370 mg/M2 by IV injection or levoleucovorin 10 mg/M2 by IV injection followed by 5-FU at 425 mg/M2 by IV injection. Repeat either regimen daily for 5 days. This 5-day course is repeated at 4-week intervals for 2 courses and then at 4- to 5-week intervals provided that complete recovery from the toxic effects of the previous course has occurred. See Dose Adjustments.
Pediatric dose
See Usual Dose.
Dose adjustments
Levoleucovorin rescue after high-dose methotrexate therapy:
Some patients may have significant but less severe abnormalities in methotrexate elimination or renal function following methotrexate administration than the abnormalities described in the preceding table. These abnormalities may or may not be associated with significant clinical toxicity. If significant clinical toxicity is observed, levoleucovorin rescue should be extended for an additional 24 hours (a total of 14 doses over 84 hours) in subsequent courses of therapy. If lab abnormalities or clinical toxicities are observed, consider the possibility that the patient is taking other medications that interact with methotrexate (e.g., medications that may interfere with methotrexate elimination or binding to serum albumin [see methotrexate monograph]). ■ Delayed methotrexate excretion may also be caused by accumulation in a third space fluid collection (i.e., ascites, pleural effusion), inadequate hydration, or renal insufficiency. Higher doses of levoleucovorin or prolonged administration may be indicated.
Inadvertent methotrexate overdose:
If the 24-hour serum creatinine has increased 50% over baseline or if the 24-hour methotrexate level is greater than 5 × 10−6 M (5 micromolar) or the 48-hour level is greater than 9 × 10−7 M (0.9 micromolar), increase the dose of levoleucovorin to 50 mg/M2 IV every 3 hours until the methotrexate level is less than 0.05 micromolar. Hydrate with a minimum of 3 liters/day and use sodium bicarbonate for urinary alkalinization (adjust to maintain a urine pH of 7 or greater).
Colorectal cancer:
In subsequent courses, adjust the dose of 5-FU based on patient tolerance of the previous treatment course. Reduce 5-FU dose by 20% for patients who experienced moderate hematologic or GI toxicity in the previous course and 30% for patients who experienced severe toxicity. 5-FU dose may be increased by 10% if no toxicity was experienced in the previous course. Levoleucovorin doses are not adjusted for toxicity.
Dilution
Levoleucovorin for injection:
Reconstitute each 50-mg vial of lyophilized powder with 5.3 mL of preservative-free NS (concentration equals 10 mg/mL). Do not use other diluents. A single dose may be further diluted immediately in NS or D5W to a concentration of 0.5 mg/mL to 5 mg/mL. Do not use if solution is cloudy or a precipitate is observed.
Levoleucovorin injection:
Available in ready-to-use single-use vials containing 175 mg (17.5 mL) or 250 mg (25 mL). Concentration equals 10 mg/mL. A single dose of these solutions may be further diluted to a concentration of 0.5 mg/mL in NS or D5W.
Filters:
Specific information not available.
Storage:
Levoleucovorin for injection (reconstituted lyophilized powder):
Store unopened vials in carton at CRT. Protect from light. Solutions reconstituted or further diluted in NS are stable for not more than 12 hours at RT. Reconstituted solutions further diluted in D5W are stable for not more than 4 hours at RT.
Levoleucovorin injection (ready-to-use solution):
Refrigerate unopened vials in carton at 2° to 8° C (36° to 46° F). Protect from light. Solutions diluted to 0.5 mg/mL in NS or D5W are stable for not more than 4 hours at RT.
Compatibility
Manufacturer states, “Due to the risk of precipitation, do not coadminister levoleucovorin with other agents in the same admixture.” Dilute only with preservative-free NS; do not use other diluents or NS with preservatives.
Rate of administration
Because of the calcium content of the solution, do not exceed a rate of injection of 16 mL/min of reconstituted solution (160 mg of levoleucovorin). When given in combination with 5-FU, flush the IV line with NS between drugs.
Actions
A folate analog. An active isomer of 5-formyl tetrahydrofolic acid, the pharmacologically active isomer of leucovorin calcium. A replacement for leucovorin calcium, which also contains the pharmacologically inactive dextro isomer. Does not require reduction by the enzyme dihydrofolate reductase. Actively and passively transported across cell membranes. Converted in vivo to the primary circulating form of active reduced folate. Levoleucovorin can counteract the therapeutic and toxic effects of folic acid antagonists such as methotrexate, which act by inhibiting dihydrofolate reductase. Terminal half-life is from 5.1 to 6.8 hours.
Indications and uses
Levoleucovorin rescue is indicated after high-dose methotrexate therapy in patients with osteosarcoma. ■ Also indicated to diminish the toxicity and counteract the effects of impaired methotrexate elimination and of inadvertent overdose of folic acid antagonists (e.g., methotrexate, pemetrexed [Alimta]). ■ Indicated for use in combination chemotherapy with 5-fluorouracil (5-FU) in the palliative treatment of advanced metastatic colorectal cancer. ■ Not indicated for the treatment of pernicious anemia and megaloblastic anemias secondary to vitamin B12 deficiency; improper use may cause a hematologic remission while neurologic manifestations continue to progress.
Contraindications
Contraindicated in patients who have had previous hypersensitivity reactions to folic acid or folinic acid.
Precautions
For IV use only; do not administer by any other route. ■ Contains calcium; limit rate of administration to no more than 16 mL/min (160 mg of levoleucovorin per minute). ■ Toxicities during combination use with 5-fluorouracil (5-FU) are similar to those observed with 5-FU alone; however, GI toxicities (particularly stomatitis and diarrhea) are observed more commonly and may be more severe or of prolonged duration. ■ Concomitant use with sulfamethoxazole/trimethoprim for the acute treatment of Pneumocystis jiroveci pneumonia in patients with HIV infection may cause treatment failure and morbidity.
Monitor:
All situations with methotrexate:
Obtain baseline serum methotrexate, serum creatinine, electrolytes, and urine pH. Monitor at least daily. ■ Adequate hydration and maintenance of a urinary pH of 7 or greater with sodium bicarbonate are indicated.
Levoleucovorin rescue after high-dose methotrexate therapy:
Patients who experience delayed early methotrexate elimination are likely to develop reversible renal failure. Continuing hydration and urinary alkalinization and close monitoring of fluid and electrolyte status are required until the serum methotrexate level has fallen to below 0.05 micromolar and the renal failure has resolved.
Inadvertent methotrexate overdose:
See Dose Adjustments; hydration with a minimum of 3 liters/day and the use of sodium bicarbonate for urinary alkalinization (adjust to maintain a urine pH of 7 or greater) may be indicated.
Combination with 5-FU:
Monitor for S/S of GI toxicity. See the 5-fluorouracil monograph for hematologic monitoring requirements.
Maternal/child:
Category C: use during pregnancy only if clearly needed. ■ Not known if levoleucovorin is excreted in human milk; has potential for harmful effects; discontinue breast-feeding.
Elderly:
Studies of levoleucovorin in the treatment of osteosarcoma did not include adults age 65 or older. ■ Dehydration, diarrhea, and severe enterocolitis have been reported in elderly patients receiving weekly levoleucovorin and 5-fluorouracil (5-FU).
Drug/lab interactions
May ameliorate the hematologic toxicity associated with high-dose methotrexate, but levoleucovorin has no effect on other established toxicities of methotrexate, such as nephrotoxicity resulting from drug and/or metabolite precipitation in the kidney. ■ Folic acid in large amounts may counteract the antiepileptic effects of phenobarbital (Luminal), phenytoin (Dilantin), and primidone (Mysoline). Seizure activity may be increased in susceptible patients. Levoleucovorin may or may not have the same effect due to shared metabolic pathways; use caution when administering concurrently with anticonvulsant drugs. ■ Small amounts of systemically administered levoleucovorin may enter the cerebrospinal fluid (CSF). Do not give levoleucovorin intrathecally; see Precautions. ■ Increases the toxicity of 5-fluorouracil (5-FU); dehydration, diarrhea, and severe enterocolitis have been reported in elderly patients receiving weekly levoleucovorin and 5-fluorouracil. ■ Concomitant use with sulfamethoxazole/trimethoprim for the acute treatment of Pneumocystis jiroveci pneumonia in patients with HIV infection may cause treatment failure and morbidity. ■ High doses of levoleucovorin may reduce the effectiveness of intrathecally administered methotrexate. ■ Levoleucovorin may enhance the toxic effects of capecitabine (Xeloda); monitor closely.
Side effects
Nausea and vomiting and stomatitis were reported most commonly. Abnormal renal function, confusion, dermatitis, diarrhea, dyspepsia, dyspnea, hypersensitivity reactions, leukopenia, neuropathy, thrombocytopenia, typhlitis (inflammation of the cecum), and taste perversion have been reported.
Combination therapy with 5-fluorouracil (5-FU):
Abdominal pain, alopecia, anorexia, dermatitis, diarrhea, fatigue, nausea, stomatitis, vomiting. Hypersensitivity reactions have been reported.
Post-marketing:
Dyspnea, pruritus, rash, rigors, temperature change.
Antidote
Keep physician informed of patient’s condition. Symptomatic treatment indicated.
Levothyroxine sodium 
(lee-voh-thigh-ROX-een SO-dee-um)
T4, l-Thyroxine
Hormone (thyroid)
Usual dose
When oral ingestion is not practical, IV dose should be 1/2 of any previously established oral dose; see Indications, Limitations of Use. Adjust in small increments as indicated. Initiate oral treatment as soon as possible.
Hypothyroidism (when oral therapy is not possible):
Usual IV starting dose would be 6.25 to 12.5 mcg/day. Increase in increments of 12.5 mcg every 2 to 4 weeks. Base dosing on clinical response and serum thyroid and TSH levels. Average maintenance dose is 50 to 100 mcg/day PO.
Myxedema coma:
300 to 500 mcg as an initial dose. Follow with once-daily intravenous maintenance doses between 50 and 100 mcg.
Pediatric dose
Given orally (may be crushed in food or liquid). See Precautions. Any IV dose should be 50% to 75% of the established oral dose. See pediatric literature for dosing guidelines.
Dose adjustments
Age, general condition, cardiac risk factors, and clinical severity of myxedema symptoms should be considered when determining the starting and maintenance dosages. ■ Reduce dose in elderly, functional or ECG evidence of cardiovascular disease (including angina), long-standing thyroid disease, other endocrinopathies, and severe hypothyroidism. ■ See Drug/Lab Interactions.
Dilution
Available in different strengths; read label carefully. Each vial of lyophilized powder is diluted with 5 mL of NS for injection (without preservatives). Shake well to dissolve completely. Reconstituted concentrations will be 20 mcg/mL for the 100-mcg vial and 100 mcg/mL for the 500-mcg vial. Do not add to IV solutions. May be given through Y-tube or three-way stopcock of infusion set.
Storage:
Store dry product at CRT and protect from light. Reconstituted solution is preservative-free and stable for 4 hours; any remaining solution is discarded.
Compatibility
Manufacturer states, “Do not add to other IV fluids.”
Rate of administration
100 mcg or fraction thereof over 1 minute.
Actions
A synthetic thyroid hormone. Effective replacement for decreased or absent thyroid hormone. Thyroid hormone synthesis and secretion is regulated by the hypothalamic-pituitary-thyroid axis. Thyroid hormones regulate multiple metabolic processes and play an essential role in normal growth and development. Actions are produced predominantly by T3 (triiodothyronine). Approximately 80% of T3 is derived from T4 (levothyroxine) by deiodination in peripheral tissues. The metabolic actions of thyroid hormones include augmentation of cellular respiration and thermogenesis, as well as metabolism of proteins, carbohydrates, and lipids. Circulating thyroid hormones are greater than 99% bound to plasma proteins (thyroxine-binding globulin [TBG], thyroxine-binding prealbumin [TBPA], and albumin [TBA]). The higher affinity of both TBG and TBPA for T4 partially explains the higher serum levels, slower metabolic clearance, and longer half-life of T4 (6 to 8 days) compared with T3 (less than 2 days). The major pathway of thyroid hormone metabolism is through sequential deiodination. The liver is the major site of degradation for both T4 and T3. Primary route of elimination is through the kidneys. Small amounts are eliminated into the bile and feces.
Indications and uses
Treatment of myxedema coma. ■ Specific replacement therapy for reduced or absent thyroid function due to any cause (usually given orally).
Limitations of use:
The relative bioavailability between oral and intravenous levothyroxine sodium for injection has not been established but has been estimated to be between 48% and 74%. Caution should be used when switching patients from oral products to the parenteral product because adequate dosing conversions have not been studied.
Contraindications
Manufacturer states, “None.” ■ Not indicated for use in treatment of obesity. Risk can outweigh benefit.
Precautions
Excessive bolus doses (greater than 500 mcg) are associated with cardiac complications, particularly in the elderly and in patients with underlying cardiac disease; see Dose Adjustments. ■ Patients with undiagnosed endocrine disorders (e.g., adrenal insufficiency, hypopituitarism, diabetes insipidus) may experience new or worsening symptoms of these endocrinopathies. Patients should be treated with replacement glucocorticoids before initiation of treatment with levothyroxine until adrenal function has been adequately assessed. Failure to do so may precipitate an acute adrenal crisis when thyroid hormone therapy is initiated as a result of increased metabolic clearance of glucocorticoids by thyroid hormone. Patients with myxedema coma should be monitored for previously undiagnosed diabetes insipidus. ■ Not indicated for treatment of obesity or for weight loss. Doses within the range of daily hormonal requirements are ineffective for weight reduction. Larger doses may produce serious or even life-threatening manifestations of toxicity, particularly when given in conjunction with sympathomimetic amines, such as those used for the anorectic effects.
Monitor:
Observe patient closely and monitor vital signs. ■ Monitor thyroid function tests (e.g., free T4 index, TSH). ■ TSH and thyroid hormone levels should be measured a few weeks after switching from IV to oral therapy. Adjust dose according to results and clinical status. ■ Monitor TSH every 6 to 8 weeks until normalized, every 8 to 12 weeks after dose changes, and every 6 to 12 months throughout therapy.
Maternal/child:
Category A: may be used during pregnancy. ■ Presumed safe in breast-feeding; use caution and observe infant. ■ Myxedema coma is a disease of the elderly. An approved oral dosage form should be used in pediatric patients for maintaining a euthyroid state in noncomplicated hypothyroidism; see Pediatric Dose.
Elderly:
See Dose Adjustments; more sensitive to effects. Atrial fibrillation is a common side effect associated with levothyroxine treatment in the elderly.
Drug/lab interactions
Addition of levothyroxine to antidiabetic or insulin therapy may result in increased antidiabetic or insulin requirements. Monitor glucose values closely, especially when thyroid therapy is initiated, changed, or discontinued. ■ Levothyroxine increases response to oral anticoagulant therapy (e.g., warfarin). Reduction in dose of oral anticoagulant may be required. Monitor PT/INR. ■ May decrease therapeutic effects of digoxin, necessitating an increase in the digoxin dose. Monitor serum digoxin levels. ■ Concurrent use of tricyclic (e.g., amitriptyline) or tetracyclic (e.g., maprotiline) antidepressants and levothyroxine may increase the therapeutic and toxic effects of both drugs, possibly due to increased receptor sensitivity to catecholamines. ■ Administration of sertraline in patients stabilized on levothyroxine may result in increased levothyroxine requirements. ■ Concurrent use with ketamine may produce marked hypertension and tachycardia. Use caution. ■ Concurrent use with sympathomimetics (e.g., epinephrine, norepinephrine) may increase the effects of sympathomimetics or thyroid hormone. May increase risk of coronary insufficiency. ■ Carbamazepine, fosphenytoin, phenytoin, and rifampin may decrease the serum concentration of thyroid hormones. Monitor levels closely. ■ Levothyroxine can increase the metabolism of theophylline, necessitating an increase in theophylline dose. Monitor theophylline levels. ■ Changes in TBG concentration must be considered when interpreting T3 and T4 values, which necessitates measurement and evaluation of unbound (free) hormone and/or determination of the free levothyroxine index. Several medications and disease states can alter TBG concentrations. See manufacturer’s prescribing information.
Side effects
Abdominal cramps, angina, arrhythmias, chest pain, diarrhea, heart palpitations, heat intolerance, increased pulse and BP, insomnia, menstrual irregularities, muscle cramps, nervousness, sweating, tachycardia, tremors, vomiting, and weight loss.
Antidote
Notify the physician of any side effect. A reduction in dose will usually decrease symptoms. Supportive treatment should be initiated as dictated by the patient’s medical status.
Lidocaine hydrochloride
(LYE-doh-kayn hy-droh-KLOR-eyed)
Lidocaine PF, Xylocaine PF, Xylocard 
Antiarrhythmic
pH 5 to 7 (Injection), 3 to 5.5 (IV infusion)
Usual dose
Ventricular arrhythmia:
50 to 100 mg as a loading dose (0.7 to 1.4 mg/kg). Repeat after 5 minutes if desired clinical response is not produced. Follow with an infusion of 1 to 4 mg/min (20 to 50 mcg/kg/min). Should not exceed a maximum dose of 200 to 300 mg during a 1-hour period.
Refractory ventricular fibrillation or pulseless ventricular tachycardia:
1 to 1.5 mg/kg of body weight (50 to 100 mg) as a loading dose. May repeat 0.5 to 0.75 mg/kg every 10 minutes to desired effect, up to a total maximum cumulative dose of 3 mg/kg/24 hr. Should not exceed 200 to 300 mg during a 1-hour period. Follow with a continuous infusion of 1 to 4 mg/min (20 to 50 mcg/kg/min).
Maintenance dose:
With return of perfusion, initiate an infusion of 1 to 4 mg/min (20 to 50 mcg/kg/min) in an average 70-kg adult. Do not exceed 4 mg/min rate. If arrhythmias occur during an infusion, give a small bolus of 0.5 mg/kg to increase plasma concentration.
Seizures unresponsive to other therapy (unlabeled):
1 mg/kg as a loading dose. If seizure does not terminate in 2 minutes, give an additional 0.5 mg/kg; a maintenance infusion of 30 mcg/kg/min has been used to prevent recurrences.
Cardiac arrest:
AHA guidelines recommend a single dose of 1 to 1.5 mg/kg. An additional dose of 0.5 to 0.75 mg/kg may be given in 5 to 10 minutes to a maximum of 3 doses or a total dose of 3 mg/kg.
Perfusing arrhythmia (stable VT, wide-complex tachycardia of uncertain type, significant ectopy):
AHA guidelines recommend doses ranging from 0.5 to 0.75 mg/kg and up to 1 to 1.5 mg/kg. Repeat 0.5 to 0.75 mg/kg every 5 to 10 minutes to a maximum total dose of 3 mg/kg. Follow with a continuous infusion of 1 to 4 mg/min (20 to 50 mcg/kg/min).
Pediatric dose
See Maternal/Child (unlabeled).
Antiarrhythmic:
AHA recommends 1 mg/kg as an IV injection followed immediately by an infusion of 20 to 50 mcg/kg/min (average of 30 mcg/kg/min). If 15 minutes have elapsed since the initial bolus dose before the infusion is started, administration of an additional bolus (1 mg/kg/min) is recommended when the infusion is initiated. Another source recommends 1 mg/kg IV. May repeat in 10 to 15 minutes × 2 if indicated. Maximum total dose is 3 to 5 mg/kg within the first hour. Follow with an infusion of 20 to 50 mcg/kg/min. Intratracheal dose suggested should be 2 to 2.5 times the IV dose.
Dose adjustments
Reduce loading dose in digoxin toxicity with AV block. ■ Consider loading dose reduction (not universally recommended) in congestive heart failure, reduced cardiac output, and liver disease. ■ Lower-end initial doses may be indicated in the elderly based on potential for decreased organ function and concomitant disease or drug therapy. ■ Reduce maintenance dose by one half in the presence of decreased cardiac output (e.g., acute MI, congestive heart failure, or shock from any cause [one source suggests not exceeding 20 mcg/kg/min]), with impaired liver function, in the elderly (over 65), and in patients receiving drugs that may decrease clearance of lidocaine or decrease liver blood flow (e.g., beta-blockers [propranolol, cimetidine (Tagamet)]). ■ Reduce maintenance dose after 24 hours or monitor blood levels; half-life of lidocaine increases with prolonged administration.
Dilution
Label must state “for IV use” and be preservative free. Bolus dose may be given undiluted.
Infusion:
Add 1 Gm of lidocaine to 500 or 250 mL of D5W (preferred), D5/1/2NS, D5NS, LR, or other compatible solutions; see chart on inside back cover. Solution gives 2 or 4 mg/mL of lidocaine. Available premixed in 0.4% or 0.8% solutions (4 or 8 mg/mL). Titrate to desired response.
Pediatric infusion:
Add 120 mg of lidocaine to 100 mL of diluent (1,200 mcg/mL). 1 to 2.5 mL/kg/hr will deliver 20 to 50 mcg/kg/min.
Storage:
Store at CRT. Discard diluted solution after 24 hours. Protect from freezing.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Because doses of lidocaine are titrated to response, additives should not be introduced into premixed solutions of lidocaine.”
One source suggests the following compatibilities:
Additive:
Not recommended by manufacturer. Physically compatible with numerous drugs. Combination may not be practical because of extensive individualized rate adjustments of lidocaine to achieve desired effects; consult pharmacist. Alteplase (Activase, tPA), aminophylline, amiodarone (Nexterone), atracurium (Tracrium), calcium chloride, calcium gluconate, chloramphenicol (Chloromycetin), chlorothiazide (Diuril), ciprofloxacin (Cipro IV), dexamethasone (Decadron), digoxin (Lanoxin), diphenhydramine (Benadryl), dobutamine, dopamine, ephedrine sulfate, erythromycin (Erythrocin), fentanyl, flumazenil (Romazicon), furosemide (Lasix), heparin, hydrocortisone sodium succinate (Solu-Cortef), nafcillin (Nallpen), nitroglycerin IV, penicillin G potassium, pentobarbital (Nembutal), phenylephrine (Neo-Synephrine), potassium chloride (KCl), procainamide (Pronestyl), prochlorperazine (Compazine), ranitidine (Zantac), sodium bicarbonate, sodium lactate, theophylline, verapamil.
Y-site:
Acetaminophen (Ofirmev), alteplase (Activase, tPA), amiodarone (Nexterone), argatroban, bivalirudin (Angiomax), cefazolin (Ancef), ceftaroline (Teflaro), ciprofloxacin (Cipro IV), cisatracurium (Nimbex), daptomycin (Cubicin), dexmedetomidine (Precedex), diltiazem (Cardizem), dobutamine, dopamine, enalaprilat (Vasotec IV), etomidate (Amidate), famotidine (Pepcid IV), fenoldopam (Corlopam), heparin, hetastarch in electrolytes (Hextend), hydrocortisone sodium succinate (Solu-Cortef), labetalol, levofloxacin (Levaquin), linezolid (Zyvox), meperidine (Demerol), micafungin (Mycamine), morphine, nicardipine (Cardene IV), nitroglycerin IV, nitroprusside sodium, palonosetron (Aloxi), potassium chloride (KCl), propofol (Diprivan), remifentanil (Ultiva), theophylline, tigecycline (Tygacil), tirofiban (Aggrastat), vasopressin, warfarin (Coumadin).
Rate of administration
Bolus dose:
25 to 50 mg or fraction thereof over 1 minute. Too-rapid injection may cause seizures.
Infusion:
Using an infusion pump delivers lidocaine in recommended doses. Adjust as indicated by progress in patient’s condition. See Dose Adjustments and the Infusion Rate chart.
| Lidocaine Infusion Rates | ||||||
| Desired Dose | 1 Gm in 500 mL Diluent (2 mg/mL) | 1 Gm in 250 mL Diluent (4 mg/mL) | ||||
| mg/min | mg/hr | mL/min | mL/hr | mg/hr | mL/min | mL/hr |
| 1 | 60 mg/hr | 0.5 mL/min | 30 mL/hr | 60 mg/hr | 0.25 mL/min | 15 mL/hr |
| 2 | 120 mg/hr | 1 mL/min | 60 mL/hr | 120 mg/hr | 0.5 mL/min | 30 mL/hr |
| 3 | 180 mg/hr | 1.5 mL/min | 90 mL/hr | 180 mg/hr | 0.75 mL/min | 45 mL/hr |
| 4 | 240 mg/hr | 2 mL/min | 120 mL/hr | 240 mg/hr | 1 mL/min | 60 mL/hr |
| Pediatric infusion: 120 mg to 100 mL diluent = 1,200 mcg/mL 1 to 2.5 mL/kg/hr = 20 to 50 mcg/kg/min | ||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


