blood (150 mL). Basic to an understanding of the pathophysiologic changes related to ICP is the Monro-Kellie hypothesis (Box 13-1). It states that the skull, a rigid compartment, is filled to capacity with essentially noncompressible contents—brain and interstitial fluid (80%), intravascular blood (10%), and CSF (in the ventricles and subarachnoid space; 10%). The blood volume is comprised of approximately 20% arterial and 80% venous blood volume. Blood volume is related to the capacitance of the vessel; when a vessel is dilated it holds more blood volume than the vessel that is constricted. The volume of these three components remains nearly constant in a state of dynamic equilibrium. If the volume of any one component increases, another component must decrease reciprocally for the overall volume and dynamic equilibrium to remain constant. If the volume of any one component increases without a reciprocal decrease in one of the other components, ICP will rise. This hypothesis applies only when the skull is fused (i.e., a closed box). Infants or very young children who have skulls with nonfused suture lines have some space for expansion of the intracranial space in response to increased volume, at least initially.
Displacement of some CSF from the ventricles and cerebral subarachnoid space through the foramen magnum to the spinal subarachnoid space and through the optic foramen to the perioptic subarachnoid space (basal subarachnoid cisterns)
Displacement of some blood by compression of the low-pressure venous system, especially the dural sinuses
Decreased production of CSF
Vasoconstriction of the cerebral vasculature, which results in a decrease in the intracranial blood volume
Displacement of CSF from the cerebral ventricular system using an intraventricular catheter (IVC) connected to an external drainage system (temporary) or a ventriculoperitoneal shunt (permanent).
Displacement of some blood by inducing arterial vasoconstriction. Vasoconstriction reduces the capacity of the artery to carry blood and thereby results in a net decrease in the volume of blood in the cranial vault.
Facilitating venous outflow from the head by elevating the head of the bed and maintaining midline head position thus promoting venous drainage through the jugular veins.
Displacement of some brain tissue volume through mechanisms to decrease intracellular volume. An example of this is seen when the increased osmotic pressure of the circulating blood draws off free water from brain cells and results in a net decrease in brain volume.
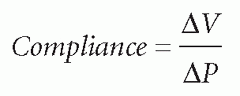
the compensatory mechanisms are exceeded, compliance is lost, and a disproportionate elevation in ICP is noted.
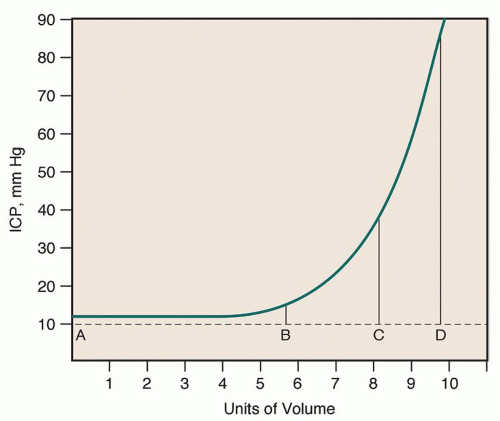 Figure 13-1 ▪ Pressure-volume curve. From point A to just before B, the intracranial pressure (ICP) remains constant, although there is addition of volume (compliance is high). At point B, even though the ICP is within normal limits, compliance begins to change, as evidenced by the slight rise in ICP. From points B to C, the ICP rises with an increase in volume (low compliance). From points C to D, ICP rises significantly with each minute increase in volume (compliance is lost). |
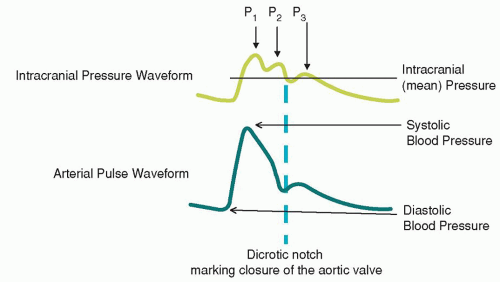 Figure 13-2 ▪ Comparison of arterial and intracranial pressure waveforms. |
P1, the percussion wave, which originates from pulsations of the arteries and choroid plexus, is sharply peaked and fairly consistent in amplitude.
P2, the tidal wave, is more variable, changes with compliance, and terminates in the dicrotic notch.
P3, the dicrotic wave, immediately follows the dicrotic notch.
situations. Because it is possible to observe responses to patient activities, treatments, and environmental factors immediately, it may be possible to use this information to develop an individualized plan of care based on avoidance of individual risk factors. ICP waveform analysis should be a focus of more clinical research to explore its ramifications for integration into clinical practice.
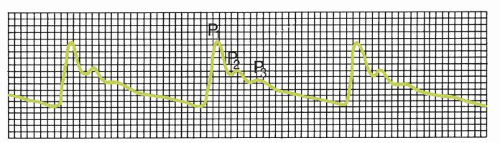 Figure 13-3 ▪ Normal intracranial pressure waveform (ICP = 8 mm Hg). |
 Figure 13-4 ▪ Abnormal intracranial pressure (ICP) waveform (ICP = 18 mm Hg). |
TABLE 13-1 ICP CHANGES RELATED TO DIFFERING PHYSIOLOGICAL CONDITIONS | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Blood pressure. The main force that maintains CBF is the pressure difference between the arteries and the veins. In the brain, cerebral venous pressure is low (approximately 5 mm Hg), so that arterial blood pressure is the most important factor in maintaining CBF. Under normal circumstances, intrinsic regulatory mechanisms maintain CBF at a constant level even with systemic arterial blood pressure changes, unless the MAP dips to less than 50 mm Hg.
Cardiac function. Systemic arterial blood pressure is dependent on cardiac output and peripheral vasomotor tone (resistance), which are primarily under autonomic control from the vasomotor center of the medulla. Cardiac arrhythmias, altered myocardial function, circulating blood volume, and cardiac disease can affect cardiac output, thus influencing CBF. In addition, various carotid sinus and aortic arch reflexes assist in maintaining a constant blood pressure. Advanced age, atherosclerosis, and certain drugs can alter these reflexes, thus affecting arterial blood pressure and CBF secondarily.
Blood viscosity. Blood viscosity is a measure of how thick the blood is. Blood viscosity is generally represented by hematocrit (Hct). A higher Hct correlates with higher viscosity. Fluid replacement therapy and anemia are two primary causes of a decrease in blood viscosity. Anemia may increase blood flow up to 30%, whereas polycythemia may decrease flow by more than 50%. When blood is viscous, vessels dilate and they constrict when blood is dilute.
Carbon dioxide (CO2), found in the blood and locally in cerebral tissue as an end product of cell metabolism, is a potent agent that influences CBF. Cerebral blood vessels respond indirectly to changes in carbon dioxide (PaCO2) levels that alter pH. Increases in PaCO2 in acidosis lead to vasodilation, thus increasing blood flow. Decreased PaCO2 leads to alkalosis and will cause vasoconstriction and result in a general decrease in blood flow. CBF generally changes by 2% to 3% for each change in PaCO2 within the range of 20 to 80 mm Hg.
Oxygen (O2) has an opposite, but less profound effect: reduction in local oxygen (PaO2) produces vasodilation by changes in pH (acidosis), and an increase in local PaO2 produces vasoconstriction. The mechanism by which this is achieved is unclear.
H+ ions are also powerful agents that influence CBF. In body fluids, CO2 combines with water to form carbonic acid, with subsequent dissociation of hydrogen ions. Hydrogen ion concentration can also be increased by lactic acid, pyruvic acid, and other acids that result from cell metabolism. Excess hydrogen ions cause cerebral vessels to dilate, which results in a net increase in CBF.
pH changes also have an effect on cerebral arterioles; a low pH (acidosis) results in vasodilation and increased CBF, and a high pH (alkalosis) results in vasoconstriction and a decreased CBF. A buildup of the metabolic end products of cell metabolism (e.g., lactic acid, pyruvic acid, carbonic acid) causes localized acidosis. An increase in the concentration of these acids will also increase CBF.
Extrinsic neurogenic control. Sympathetic innervation comes from postganglionic fibers of the superior cervical sympathetic ganglion that innervate the carotid and vertebral arteries and major intracranial branches. Norepinephrine, a vasoconstrictor, is released from the sympathetic fibers. Parasympathetic fibers come from the facial and superficial petrosal nerves to innervate large- and small-diameter cerebral blood vessels. They use acetylcholine as a neurotransmitter, which causes vasodilation.
Intrinsic neurogenic control. The intrinsic pathways originate in the brainstem and interneurons in the cerebral cortex. These pathways originate in the brainstem from the locus caeruleus (neurons use norepinephrine to produce microcirculatory vasodilation), raphe nuclei (neurons use serotonin, a vasoconstrictor), and fastigial nuclei of the cerebellum. Cortical interneurons contain both vasoconstrictor and vasodilator substances.
70 mm Hg in adults. In the pediatric population, thresholds are less well defined and most likely depend on the age of the patient. Current recommendations for the pediatric patient are to maintain CPP between 40 and 65 mm Hg.
In the center of the ischemic area is a core of dead or dying cells surrounded by an area of minimally surviving cells. This area, called the ischemic penumbra (halo around the infarction), represents the cells at greatest risk for neuronal death. The cells of the penumbra receive marginal blood flow resulting in altered metabolic activities. However, these cells are still alive and salvageable.
Local autoregulation, responsiveness to chemical-metabolic factors, and perfusion pressure are impaired or lost.
An inadequate supply of oxygen and glucose causes a switch to anaerobic metabolism, a decrease in ATP production, and an increase in lactate production.
A decrease in ATP production leads to ineffective cellular function and dysfunction of ATP-dependent neurotransmitter reuptake.
Ischemic neurons release excessive amount of the excitatory neurotransmitter glutamate; this results in increased neuronal necrosis.
Glutamate binds to N-methyl-D-aspartate (NMDA) receptors that are normally blocked by magnesium; this causes increased cell permeability to sodium and calcium ions.
Cellular swelling results primarily from the increased influx of sodium ions.
Cell lysis results from cellular swelling intracellular calciumactivated processes that lead to cell death.
Cellular lysis releases calcium ions into the postsynaptic neuron causing further neuronal ischemia and the release of more glutamate, which binds to NMDA receptors and increases cell membrane permeability to sodium and calcium, and the cycle of programmed cell death is perpetuated.
The increase in intracellular calcium levels activates phospholipases and proteases, which generate oxygen-free radicals and nitric oxide. This leads to membrane, mitochondrial, and microtubular cellular damage and eventual death.
This cell death results in the release of glutamate, which binds to NMDA receptors and increases cell membrane permeability to sodium and calcium, and the cycle of programmed cell death is perpetuated.
Conditions that increase brain volume
Space-occupying masses (e.g., hematomas, abscesses, tumors, aneurysms)
Cerebral edema (e.g., brain injuries, Reye’s syndrome)
Conditions that increase blood volume
Obstruction of venous outflow
Hyperemia
Hypercapnia
Cerebral artery vasodilation
Conditions that increase CSF volume
Increased production of CSF (e.g., choroid plexus papilloma)
Decreased absorption of CSF (e.g., communicating hydrocephalus, subarachnoid hemorrhage [SAH])
Obstruction to flow of CSF (e.g., noncommunicating hydrocephalus)
increased CBF. This, in turn, may lead to increased edema and ICP. This circular sequence continues until the autoregulatory mechanisms are inactivated. CBF passively responds to arterial blood pressure. CBF and CPP cannot be maintained in relationship to rising ICP. The CPP approaches zero and the CBF ceases. Blood vessels and brain tissue are compressed, and herniation and death follow.
Falx cerebri is a double fold of dura mater that drops into the longitudinal fissure and is partially responsible for dividing the supratentorial space into the left and right hemispheres.
Tentorium cerebelli is a double fold of dura mater that forms a tent-like partition (higher in the middle) between the cerebrum and cerebellum. The area above the tentorium is the supratentorial space, and the area below the tentorium is the infratentorial space. To allow the brainstem, blood vessels, and accompanying nerves to pass through the tentorium, there is an oval opening in the tentorium called the tentorial notch or incisura.
Falx cerebelli is a double fold of dura mater that separates the cerebellum (in the infratentorial space) into left and right sides.
a lesion located on the central neural axis (diencephalon).
an extracerebral lesion located around the central apex of the cranium (midline).
bilateral lesions in the hemispheres.
unilateral cingulate herniation.
TABLE 13-2 FACTORS ASSOCIATED WITH INCREASED INTRACRANIAL PRESSURE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
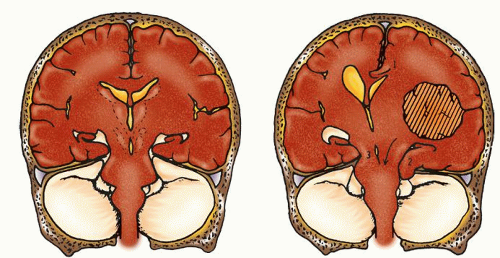 Figure 13-5 ▪ Cross-section of a normal brain (left) and a brain with intracranial shifts from supratentorial lesions (right). (1) Herniation of the cingulate gyrus under the falx. (2) Herniation of the temporal lobe into the tentorial notch. (3) Downward displacement of the brainstem through the notch. (From: Plum, F., & Posner, J. (1972). Diagnosis of stupor and coma. (2nd ed.). Contemporary neurology series. Philadelphia: F.A. Davis.) |
Deterioration in the level of consciousness (LOC) (confusion and restlessness)
Bilateral small, reactive pupils (in early diencephalic stage)
Gradual loss of upward (vertical) gaze
Contralateral monoplegia or hemiparesis to hemiplegia
TABLE 13-3 PROGRESSION OF SIGNS AND SYMPTOMS OF THE CENTRAL SYNDROMEa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
recovery. However, after the area of pathophysiology has expanded beyond the diencephalon and into the brainstem, the process is generally irreversible and prognosis poor. The underlying pathophysiology is that ischemia and compression, both reversible conditions, are the basis for the signs and symptoms of diencephalic involvement. Once the midbrain becomes involved, infarction has begun and the condition is most likely irreversible.17
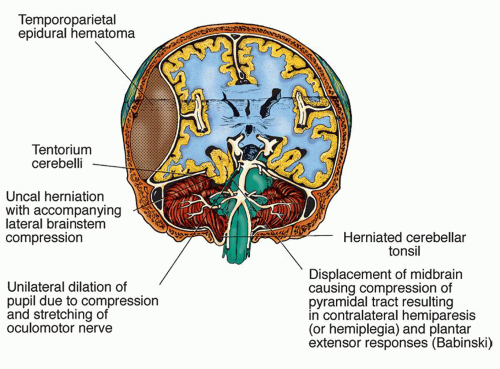 Figure 13-6 ▪ Cross-section of the brain showing herniation of part of the temporal lobe through the tentorium as a result of a temporoparietal epidural hematoma. (From: Kentzel, K. C. (1997). Advanced concepts in clinical nursing (2nd ed.). Philadelphia: J. B. Lippincott.) |
Gradual ipsilateral (to primary lesions) pupillary dilation, sluggish pupillary reaction to light (earliest sign), and possible development of an ovoid pupil (CN III)
Paralysis of the oculomotor extraocular muscles (CN IV, VI)
Restlessness, then LOC deteriorating from stupor to coma (frontal lobe and diencephalon)
Contralateral hemiparesis or hemiplegia (motor strip of frontal lobe and cerebral peduncles)
Most often, progression to decerebrate posturing; note that decorticate posturing is unusual (red nucleus lesion)
Positive Babinski’s signs (upper motor neuron lesion)
Respiratory changes (e.g., hyperventilation) (pontine lesion)
Finally, dilated, fixed pupils; flaccidity; and respiratory arrest (brainstem infarction)
TABLE 13-4 PROGRESSION OF SIGNS AND SYMPTOMS OF THE UNCAL SYNDROMEa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Direct compression of the brainstem, cerebellum, or their vascular supply
Upward transtentorial herniation of the brainstem and cerebellum through the tentorial incisura, resulting in maximal pressure on the midbrain
Downward herniation of one or both cerebellar tonsils through the foramen magnum to the cervical spine with compression of the medulla, an immediate cause of death (Fig. 13-7)
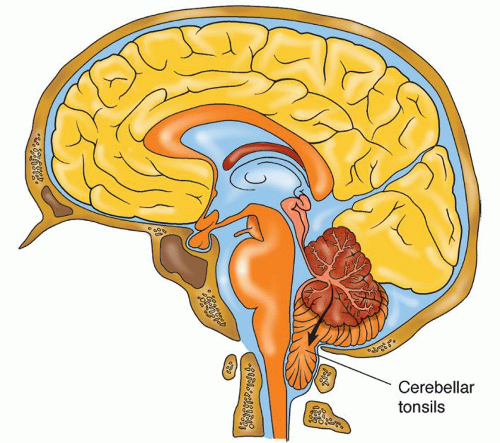 Figure 13-7 ▪ Herniation of the cerebellar tonsils into the foramen magnum is the final outcome of increased intracranial pressure. Respiratory centers within the medulla oblongata are compressed, and apnea leads to cardiac arrest and death. |
Immediate onset of deep coma
Conjugate downward deviation of the eye to failure of upward voluntary or reflex movement (pretectal compression)
Small, equal, fixed pupils (pontine compression) to unequal, midpoint fixed pupils
Decerebration
Abnormal respiratory patterns (e.g., slow rate with intermittent deep sighs or ataxia)
Vital sign abnormalities (Cushing’s signs)
CHART 13-1 Signs and Symptoms of Impending Herniation | ||
|---|---|---|
|
A rising systolic pressure widening pulse pressure
Bradycardia
Irregular respirations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


