2CF Co-ordinatory/Senior Research Physiotherapist, Great Ormond Street Hospital for Children
3Children’s Respiratory/Community Nurse Specialist, Nottingham Children’s Hospital
- understand the inheritance of primary ciliary dyskinesia and cystic fibrosis
- be familiar with the pathophysiology of the conditions
- understand the approach used to diagnose these conditions
- appreciate the importance of early diagnosis and pre-emptive treatment
- understand the range of pulmonary pathogens seen in these conditions and the approach to their treatment
- appreciate the importance of multidisciplinary working, including physiotherapy, nursing management and national standards of care.
Chapter 10 provided an insight into asthma treatment and management. The evidence suggests that there is a strong family link, so we will now move on to look at two other genetically inherited respiratory disorders – primary ciliary dyskinesia and cystic fibrosis.
Primary ciliary dyskinesia
In order to maintain healthy lungs which are free of infection, inhaled particles and micro-organisms must be removed from the respiratory tract. The most effective mechanism for achieving this is the mucociliary escalator. The respiratory tract is lined with cells which have tiny ‘hairs’, or cilia, on their surface. These cilia move in a co-ordinated fashion, like a ‘Mexican wave’ at a football match, and waft micro-organisms and other particles up the respiratory tract to the larynx, where fluid and particles are eventually swallowed. In primary ciliary dyskinesia (PCD), the cilia either fail to move at all or they move in an unco-ordinated fashion. As a result micro-organisms are not cleared from the airways and infection occurs.
Primary ciliary dyskinesia occurs in between 1/15,000 and 1/30,000 live births. Most cases are autosomal recessive and the condition is more prevalent in communities where consanguinity is common, notably the Asian community in some parts of the UK (O’Callaghan et al. 2010).
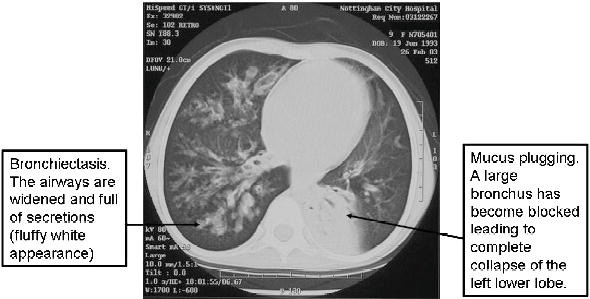
Individuals with PCD, as with other types of lung disease where there is chronic infection, are at risk of developing bronchiectasis. This is defined as an irreversible widening of the small airways. This interferes with the mucociliary escalator and allows secretions to accumulate and become secondarily infected.
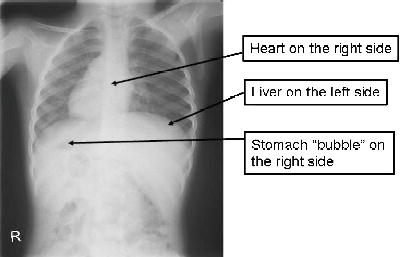
Approximately half of patients with PCD have dextrocardia (heart on the right side of the chest) and situs inversus (internal organs such as the lungs, liver, spleen and stomach are on the opposite side) (Bush et al. 2007) (Figure 12.1). This occurs because the cilia are responsible for arranging the developing organs in their correct location in the embryo. However, most babies with situs inversus do not have PCD (Bush et al. 2007). Many patients with PCD are diagnosed late, sometimes after lung damage has occurred. The mean age at diagnosis was between 4 and 5 years in one series (Coren et al. 2002). Apart from situs inversus, key clinical clues for PCD are:
- persistent nasal discharge, starting in the newborn period
- respiratory distress (fast laboured breathing) in the newborn period
- a persistent wet cough in older children
- chronically discharging ears (often following grommet insertion).
The diagnosis of PCD is undertaken in a small number of specialised centres in the UK (currently Leicester Royal Infirmary, the Royal Brompton and Southampton University Hospitals NHS Trust). A measurement of the nitric oxide levels at the nose is sometimes used as a screening test (Karadag et al. 1999). (Nitric oxide levels are abnormally low in PCD.) However, the definitive test requires samples of respiratory epithelial cells with cilia, which are taken from the nose using a tiny brush and are examined under the microscope to determine how quickly the cilia beat and whether they do this in a co-ordinated fashion (Stannard et al. 2010). The cilia are also examined using an electron microscope to see if their structure looks abnormal (Stannard et al. 2010).
Much of the treatment of PCD is not based on properly designed clinical trials but rather is borrowed from the treatment used for children with conditions such as cystic fibrosis and asthma. In PCD, the basis of treatment is close monitoring of children, including regular cough swab or sputum samples for microbiology, physical examination and measurement of pulmonary function. Children should have daily chest physiotherapy to help clear respiratory secretions. In young children this will be by percussion and postural drainage (‘bash and tip’) whereas older patients will use positive airway pressure techniques such as the positive expiratory pressure (PEP) mask or controlled breathing and coughing (active cycle of breathing). These techniques are discussed in more detail in the section below on cystic fibrosis.
Respiratory infection may be detected in cough swab and sputum samples, or suspected when there is a troublesome wet cough. In both instances, children should be treated with antibiotics which may need to be given intravenously in severe cases. The most common organisms found are Staphylococcus aureus, Haemophilus influenzae and Pseudomonas aeruginosa. Therefore annual influenza immunisation should be given. Children are also vulnerable to mucus collection in the middle ear (otitis media with effusion) which can affect hearing and the development of speech. Insertion of grommets should be avoided as this can lead to persistent discharge. The problem usually resolves in the teenage years (Bush et al. 2007). Nasal discharge can be treated by washing out with a ‘douche’ device (Bush et al. 2007), so in the long term children treated with this approach do well and lung function is better preserved than in patients first diagnosed in adult life (Ellerman and Bisgaard 1997).
Cystic fibrosis
Pathophysiology
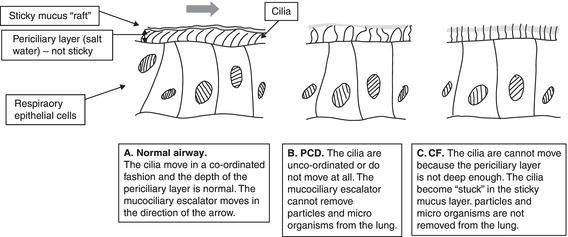
Like PCD, lung disease in cystic fibrosis (CF) occurs because of failure of the mucociliary escalator. However, the mechanism is very different. Figure 12.2 shows how the cilia function in the normal lung, compared to PCD and CF. The periciliary layer can be likened to a layer of lubricant in a machine. If the layer is not thick enough the machine cannot run smoothly.
In CF the periciliary or ‘lubricant’ layer is not as deep as it should be. This layer is composed of salt water. In CF the channel which allows chloride ions to pass out of the cell is not working, whereas the sodium channel, which allows sodium to pass into the cell, is overactive. This means that there is not enough salt (sodium chloride) in the periciliary layer and hence not enough water is drawn in through osmosis. While the periciliary layer allows the cilia to move freely, the mucus is stickier and the cilia become stuck. The mucociliary escalator does not work and particles and micro-organisms build up in the lung. This leads to secondary infection and plugging of small airways with mucus, illustrated in Figure 12.3.
Genetics
Cystic fibrosis is an autosomal recessive condition, which means an individual must have two defective forms of the CF gene to have the condition. In the general population in the UK, 1/25 individuals is a carrier; the CF gene is located on chromosome 7 (Smyth 2008). There are over 1000 different mutations of the CF gene which can cause disease. However, in the UK, one mutation, δ F508, is very common; 50% of UK CF patients have two copies of this gene and 90% have at least one copy. The normal CF gene is responsible for making the chloride channel (cystic fibrosis transmembrane conductance regulator or CFTR) which maintains the periciliary layer at the correct thickness and hence allows the mucociliary escalator to function. Mutations or defects of the CF gene do not make functioning CFTR. Some mutations of CFTR (such as R117H) are referred to as ‘dominant mild mutations’. When one of these genes is present, the CF disease may be less severe.
Diagnosis
Antenatal diagnosis
Close liaison between the clinical genetics and CF teams is essential. If a couple have had a previous baby with CF, and both parents have been genotyped, then chorion villus sampling may be performed in the first trimester to allow genotyping of the fetus. Some parents may opt for termination if CF is diagnosed (Polnay et al. 2002). The first manifestation of CF may be on the 16-week ultrasound scan where echogenic bowel or bowel dilation may be seen. Where both these signs are present there is around a 17% chance of the fetus having CF (Muller et al. 2002). At this point, both parents should have their CF genotype determined and (if the parents wish to proceed) amniocentesis should be performed, allowing genotype testing of the fetus.
Newborn screening
In the UK every newborn infant is now screened for cystic fibrosis and a number of other genetic disorders (UK Newborn Screening Programme Centre 2010). On or around the fifth day of life, a dried blood spot is collected by means of a heel prick,and sent for immune reactive trypsin (IRT) testing (UK Newborn Screening Programme Centre 2010). IRT, an enzyme secreted by the pancreas, is raised in newborns with CF, due to early pancreatic damage. If the IRT is raised, then blood from the same sample is analysed for the four most common CF mutations. If two CF mutations are found, then the child is referred to a paediatrician. If only one mutation is found then further analysis of the same specimen is done, looking for around 30 further mutations. If no further mutations are found then a new blood sample is collected and the IRT test is repeated. If still raised, the child is referred. If no mutations were detected and yet the original IRT was very high, then the IRT is repeated and the child referred if the IRT is still raised. Screening is highly accurate, missing only around one infant in 40,000 screened. Of those missed, many will have meconium ileus (see below) and so will be diagnosed clinically.
Figure 12.3 A high-resolution CT scan of a 10-year-old girl with CF showing bronchiectasis and the effects of mucus plugging.
The sweat test
The sweat test is the gold standard for diagnosis in CF (Multidisiplinary Working Group 2003). Sweating is induced using the chemical pilocarpine, applied to the smooth surface of the forearm. An electrical current is used to help the pilocarpine enter the skin. The sweat is then collected over a period of 30 min, either using filter paper or in a special device called a Macroduct® (Westcor Biomedical Systems, Utah, US). The collected sweat is then weighed and the sweat chloride level measured.
- >60 mmol/L Indicates CF
- 40–60 mmol/L Suspicious – further testing and careful follow-up needed
- <40 mmol/L Normal
Clinical features and complications
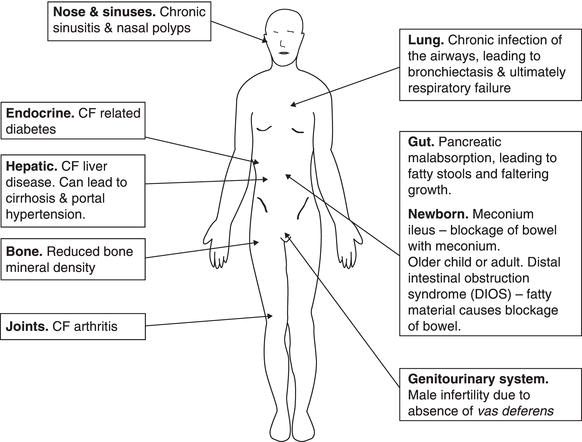
Although we have focused on lung disease so far in this discussion, CF is a multisystem disease. Figure 12.4 shows the effects of CF on other body systems.
Gastrointestinal problems
Meconium ileus
Approximately 15% of babies with CF will come to medical attention, shortly after birth, through failure to pass the first black sticky stool (meconium). This can lead to abdominal distension and vomiting. Treatment is by instilling radiological contrast into the baby’s rectum and upward to the level of the obstruction – usually the terminal ileum. The pressure of the radiological contrast may shift the obstruction and x-rays may be taken at the same time to determine if the colon is small (‘microcolon’). If this procedure is unsuccessful or if a perforation of the bowel occurs, then surgery with formation of an ileostomy will be needed. In these circumstances, a period of parenteral nutrition may be essential. Both the use of parenteral nutrition and the occurrence of meconium ileus increase the chances of subsequent CF liver disease (see below).
Malabsorption and growth faltering
Most babies diagnosed with CF by newborn screening will already have faltering growth at the time they are referred with a positive screening test. The problems of sticky secretions and obstruction seen in the lungs also occur in the pancreas, leading to a deficiency of pancreatic enzymes and bicarbonate. This means that food (initially milk) is not digested and absorbed, particularly the calorie-rich fats. The same applies to the fat-soluble vitamins so supplements of vitamins A and E are given routinely. Vitamins D and K are also given if deficiency is shown or (in the case of vitamin K) there are problems with blood clotting. The enzyme deficiency is overcome by giving enzyme supplements. These can be broken down by acid in the stomach and so enzymes are given as enteric-coated granules. Older children take these in capsules. Younger children and infants will have granules; in the case of babies in the first few weeks of life, these enzyme granules are given with a small amount of fruit puree. The amount of enzyme given is determined by how well the baby grows and the appearance of the stools. Mothers who wish to breastfeed their baby with CF should be encouraged to do so.
In some children malabsorption is difficult to control with enzymes and neutralising gastric acid with drugs such as omeprazole may help. Children with CF also burn more energy (some of this is due to lung disease and infection) and it is recommended they have 120–150% of the recommended dietary intake of calories. In some cases dietary supplements or gastrostomy feeding may be necessary.
Distal intestinal obstruction syndrome
Pancreatic enzyme supplements are not perfect and some of the fatty component of food may not be fully digested, even when the enzyme capsules are taken as prescribed. Children and young people with CF may develop abdominal pain and difficulty passing stool, due to a collection of partially digested food at the narrowest point in the bowel (the ileocaecal valve). This is termed distal intestinal obstruction syndrome. Milder cases respond to laxatives such as lactulose. In more severe cases the radiological contrast medium gastrograffin is given orally.
Respiratory problems
Although CF is a multisystem disorder, most people with CF will die from respiratory failure. Prognosis has improved greatly in the last four decades and life expectancy for babies born in the last decade is around 40 years. This is expected to improve further in future.
In CF there is a vicious circle of infection, inflammation and lung damage which leads to bronchiectasis and ultimately to respiratory failure. Detecting and treating infection early is of paramount importance, if children are to achieve the best possible life expectancy and quality of life. Children with CF admitted to hospital for treatment of respiratory infection may look well compared to the child in the next bed with acute severe asthma. Both are having life-saving treatment – the only difference is that the child with CF sees the benefit of this treatment some years hence.
Treatment
Diagnosis and treatment of infection
As with PCD, a cough swab or sputum sample should be taken at every visit and the child seen at least every 2 months. One of the first organisms found in infants and younger children is Staph. aureus and children up to 3 years of age are commonly given prophylactic antistaphylococcal antibiotics (Smyth and Walters 2003). Infection with H. influenzae is also common in young children and usually responds to oral antibiotics. More problematic is infection with P. aeruginosa. This organism causes chronic infection, is resistant to many antibiotics and may become more resistant as infection in an individual progresses. The key to successful treatment is to eradicate the organism as soon as it is detected. In the UK a combination of the antibiotics ciprofloxacin (orally) and colistin (nebulised) is frequently given, often for 3 weeks initially though courses may need to be as long as 3 months (Langton Hewer and Smyth 2009). In the US nebulised tobramycin (often for 1 month) may be used on its own (Langton Hewer and Smyth 2009). In some children, eradication fails and infection with P. aeruginosa becomes chronic. Infection may be controlled (though not eradicated) with long-term treatment with nebulised colistin and intravenous antibiotics given for periods of worsening cough and breathlessness. As with PCD, annual influenza immunisation should be given.
Physiotherapy
As well as antibiotic treatment, chest physiotherapy is crucial to keeping children with CF well, as with PCD. This will be discussed later.
Other respiratory treatment
As the basic problem causing CF lung disease is failure of the mucociliary escalator, it seems sensible to use drugs designed to restore this function. Hypertonic saline (6% or 7%) is given twice daily by nebuliser and this can be shown to restore the function of the mucociliary escalator (Donaldson et al. 2006). Over a period of a year, it reduces by half the number of exacerbations of chest symptoms which patients experience (Elkins et al. 2006a).
Dornase-α is a form of DNase, an enzyme naturally produced by the body to break down DNA. Sputum contains DNA from dead neutrophils (cells that fight bacterial infection) and this makes the sputum sticky. Nebulised dornase-α, given once daily, leads to a small but important improvement in lung function which is maintained for up to 2 years (Jones et al. 2003).
Azithromycin is an antibiotic which, whilst it does kill P. aeruginosa, is beneficial in patients with CF who have chronic infection. Over a period of a year it leads to increased weight in CF patients and to fewer exacerbations (again reduced by around 50%) (Saiman et al. 2003). It is effective when taken three times per week.
Steroid treatment (mainly prednisolone) is used sparingly in CF as long-term use leads to unacceptable side-effects (Cheng et al. 1999a). Its main use in current practice is to treat an allergic response to the airborne mould Aspergillus fumigatus (allergic bronchopulmonary aspergillosis).
For advanced lung disease, children may need home oxygen, either at night or continuously, most conveniently given with an oxygen concentrator and cylinders. Referral for lung transplantation should be made according to local guidelines (commonly when the forced expiratory volume in 1 s (FEV1) is <30% predicted), and will be discussed further in Chapter 13 (Kerem et al. 1992). Non-invasive ventilation using a nasal or facemask may be helpful at this stage and should be started sooner rather than later, but this is discussed in more detail in Chapter 7.
Nose and sinuses
The respiratory tract includes the nose and paranasal sinuses. The problems of build-up of secretions and secondary infection also occur here, leading to chronic sinusitis in some patients. This may be associated with the formation of fleshy lumps of inflammatory tissue in the nasal passages, called nasal polyps. These respond (after some weeks) to nasal steroid treatment. Occasionally surgery is needed but the polyps tend to recur.
Liver disease
The smaller bile ducts may become blocked with sticky bile in CF, leading to back-pressure and scarring of the liver, a picture known as centrilobular fibrosis. This can cause progressive damage to the liver, leading to cirrhosis – islands of normal liver surrounded by fibrous ‘pockets’ of scar tissue. This may cause back-pressure in the main blood vessel leading from the gut to the liver (the hepatic portal vein) which leads to ‘blisters’ in the blood vessels around the oesophagus (oesophageal varices). This can cause life-threatening bleeding, while the damage to the liver cells can prevent the production of adequate amounts of clotting factors, exacerbating the problem. The incidence is just under 2% per 100 patients per year and there is a sharp fall in the risk of liver disease in CF after 10 years of age (Colombo et al. 2002). Screening for CF liver disease is with annual liver function tests and liver ultrasound where the tests abnormal. At the first sign of early liver disease, treatment should be commenced with ursodeoxycholic acid and continued life long (Cheng et al. 1999b). Where liver disease is progressive, referral should be made to a liver unit.
Cystic fibrosis-related diabetes
Just as secretion of enzymes from the pancreas can be impaired, so too production of insulin from the islet cells may reduce over time in people with CF. This can lead to CF-related diabetes which is present in between 12% and 15% of people with CF with an average age of onset of 20 years (Brennan et al. 2004). CF-related diabetes is different from type 1 diabetes in that patients rarely present with drowsiness, acidosis and dehydration (diabetic ketoacidosis). Rather, CF-related diabetes comes on slowly, with poor weight gain or weight loss in older patients. The symptoms may be insidious and so annual screening for CF-related diabetes from the teenage years is essential. This is usually with an annual oral glucose tolerance test. Glucose is given orally with blood glucose measured immediately beforehand and after 2 h. In CF the 2-h level is characteristically raised. In most cases, treatment is with a long-acting insulin analogue such as glargine, which can be administered once daily.
Bones and joints
Cystic fibrosis patients are at risk of reduced bone mineral density because of malabsorption of vitamins D and K, reduced weight-bearing activity, oral steroid use and the effects of chronic infection. From the teenage years, screening should be done every 2 years using dual-energy x-ray absorptiometry. Treatment with vitamin D and calcium is sensible but not of proven benefit. Drugs such as alendronate may improve bone mineral density but have side-effects (such as bone pain) and little is known about their long-term effects (Conwell and Chang 2009).
A specific form of arthritis may be troublesome in patients with CF, often flitting from one joint to the next. This does not cause joint damage and usually responds to simple pain relief. In addition, the frequent use of ciprofloxacin may be associated with joint pain and if this occurs, the antibiotic should be stopped, where possible.
Fertility
Discussions about fertility should commence during adolescence, as part of an annual review (Lyengar and Coleman 2005). Most men with CF are infertile, due to agenesis of the vas deferens (the tube which carries sperm from the testis). However, sexual health advice should still include condom use. Men with CF can father children with specialised fertility treatment. Women with CF, contrary to many older textbooks, have normal fertility, provided they are not severely unwell and their lung function is stable.
The best predictors for pregnancy outcome in women with CF are FEV1 and Body Mass Index (BMI). There is an increased risk of mortality with pregnancy for women with CF who have a decline in lung function, weight and general nutritional status (Lyengar and Coleman 2005). Therefore preconceptual counselling should involve a discussion about the impact of pregnancy on a woman’s general health.
Some of the issues that women with CF may face with pregnancy include adapting physiotherapy to a growing ‘bump’, life issues, i.e. living long enough to see their child grow up, and ultimately general wellbeing (Goddard and Bourke 2009). Goddard and Bourke (2009) also emphasise the need for caution when advising CF women about pregnancy but at the same time respecting their wishes.
Standards of care
It is clear from the above description of multisystem disease that effective care must be multidisciplinary, with a dedicated team comprising a doctor, specialist nurse, physiotherapist, dietician, social worker, psychologist, pharmacist, teacher, play specialist and administrative support. A number of important ‘standards of care’ documents have been produced with guidance for those providing and commissioning care (Cystic Fibrosis Trust 2001a,b, 2011; Kerem et al. 2005). There is an increasing and appropriate emphasis on quality improvement and recording patient data, confidentially, on national registries such as those in the US and the UK. These registries can measure improvements in survival year on year, allow comparison of outcomes between centres and identify patients who may be suitable to take part in clinical trials.
For both primary ciliary dyskinesia and cystic fibrosis, early diagnosis and timely identification of complications are essential. Careful multidisciplinary management will achieve the best outcome in terms of quality of life and survival. Children are often expected to undergo multiple therapies and any new treatment must be subject to a rigorous comparison with existing therapy so that children have a rational regimen which is as simple as possible. Therefore multidisiplinary working is paramount, including nursing support and care in the community to assist parents with simplifying treatment.
Physiotherapy and promoting independence
As discussed in Chapter 1, anatomical and physiological differences between the respiratory systems of children and adults have important consequences for the physiotherapy management of children with respiratory disease, in terms of assessment, treatment and choice of techniques. Assessment and treatment of young children, teenagers and young adults require skilful age-appropriate communication with the child, the family and within the multidisciplinary team.
It is essential to include the child, parents, relatives and carers as part of the care team. As age increases, the older child and then the teenager/young adult can be progressively more involved in the decision-making process. Children’s awareness of the implications of illness and treatment develops as they grow older and they should be encouraged to take on more responsibility for their treatment. Teenagers, particularly, have a more sophisticated understanding and may be beginning to think about the future and the impact of chronic illness on school, social life and body image.
Respiratory assessment and examination
Assessment
Careful assessment is essential to identify problems requiring physiotherapy intervention. It is based on both a subjective and objective assessment and the results dictate the formulation of an appropriate treatment plan. For those requiring long-term intervention, regular assessment is required to evaluate the ongoing effectiveness of the treatment in relation to problems and goals (Middleton and Middleton 2008).
Discussion with medical staff, nursing staff and the parents/carers is essential to gain information about recent changes. In the acute situation, it is particularly important to ascertain how stable the child is: their ability to tolerate handling, the timing and modality (oral, nasogastric or intravenous route) of feeds and whether the child is sufficiently rested to tolerate a physiotherapy treatment if it is appropriate. Results of investigations and other relevant observations should be referred to as appropriate and relevant information gleaned from the observations charts as usual.
In chronically ill children who require home physiotherapy, good communication between the specialist and primary healthcare teams is essential.
Examination
Examination of the older child is similar to that of the adult. Other relevant observations such as the behaviour of a child can often give important clues about their respiratory status. Agitation or irritability may be a sign of hypoxia, while the child in severe respiratory distress may be withdrawn and lie completely still. It is also important to note muscle tone as there may be an increase in work of breathing and difficulty with coughing and expectoration in those with hypotonia. Hypertonia may also be associated with difficulty in clearing secretions. Abdominal distension may cause or exacerbate respiratory distress, as the diaphragm is placed at a mechanical disadvantage.
Airway clearance
Removal of bronchial secretions is considered to be the primary aim of airway clearance techniques. In the acute situation retained secretions may lead to airway obstruction and atelectasis. In chronic disease such as CF, recurrent infection and inflammation result in damage and destruction of the airways and the mucociliary escalator, as discussed previously; also, retained secretions cause an increase in airway resistance and obstruction.
Autogenic drainage (AD)
High-frequency chest wall oscillation (HFCWO)
Intrapulmonary percussive ventilation (IPV)
Oscillating positive expiratory pressure:
- Flutter®
- R-C Cornet®
Positive expiratory pressure (PEP)
High positive expiratory pressure
Postural drainage and percussion
Over the past three decades several different airway clearance techniques (ACT) have been developed, all of which are reported to enhance mucus clearance and are advantageous in that they enable older children and adults to be independent in performing their treatment (Box 12.1). The general aim of all airway clearance techniques is to increase lung volume and enhance airflow. Some may also affect the rheological properties of mucus.
The majority of airway clearance studies have been undertaken in cystic fibrosis, where no single technique has been identified as being superior to others (Elkins et al. 2006b; Main et al. 2005; Morrison and Agnew 2009; van der Schans et al. 2000). A few other comparative studies have been undertaken in non-CF bronchiectasis but again have found little difference between the techniques investigated (Patterson et al. 2005, 2007).
Impaired cough as a consequence of weakness from neuromuscular disease (e.g. Duchenne muscular dystrophy and spinal muscular atrophy) can cause serious respiratory complications, including atelectasis, pneumonia, airway obstruction and acidosis (Miske et al. 2004). Chronic respiratory insufficiency and respiratory failure will ultimately result from chronic weakness of respiratory muscles, shallow breathing and ineffective cough. In these situations, independently performed airway clearance techniques may not be feasible but options such as the mechanical insufflation/exsufflation device (‘cough assist’) and other non-invasive forms of positive pressure ventilation appear to be safe and well tolerated, with a growing body of evidence to support their efficacy (Chatwin and Simonds 2009; Fauroux et al. 2007; Panitch 2006; Vianello et al. 2005).
Autogenic drainage
Autogenic drainage consists of a three-phase breathing regimen which can be performed in sitting or an appropriate postural drainage position (Schöni 1989). Mucus clearance is facilitated by the adjustment of tidal volume breathing during which the highest possible expiratory airflow is reached without causing airway closure. The three phases consist of a period of breathing at low lung volume when secretions are mobilised from the peripheral areas, followed by breathing at mid lung volume to ‘collect’ the mobilised secretions, and high lung volume breaths to clear and expectorate (IPGCF 2009; Pryor and Prasad 2008; Schöni 1989). Autogenic drainage should only be taught by an experienced therapist, skilled in the technique.
Active cycle of breathing techniques (ACBT)
The ACBT is a flexible breathing regimen consisting of breathing control, thoracic expansion exercises and the forced expiration technique (Pryor and Prasad 2008; Pryor et al. 1979). Breathing control is quiet, gentle breathing at the patient’s own comfortable rate, keeping the upper chest and shoulders relaxed, and is used between the more active parts of the cycle to prevent any increase in airflow obstruction or tiredness. Thoracic expansion exercises are deep breathing exercises emphasising inspiration with a quiet and relaxed expiration. These exercises help to loosen bronchial secretions based on the concept of interdependence and increased collateral flow. The forced expiration technique consists of one or two ‘huffs’ interspersed with periods of breathing control.
High-frequency chest wall oscillation
High-frequency chest wall oscillation (sometimes know as high-frequency chest wall compression or vest therapy) has been used in the USA for many years but is now becoming more widely available. The equipment consists of an electric air compressor which is connected to an inflatable jacket which fits snugly over the thorax. The air pulse generator delivers intermittent positive pressure (pulses of air) into the jacket, which vibrate the chest wall at oscillatory frequencies of 5–20 Hz. Mucus clearance is facilitated by the airflow oscillation and vibration of the airway walls (Pryor and Prasad 2008; Warwick et al. 2004). A total treatment time of 20–30 min is usually recommended and nebulised hypertonic saline is often used during treatment, which is interspersed by episodes of huffing and coughing.
Intrapulmonary percussive ventilation
The intrapulmonary percussive ventilation (IPV) device consists of a high-flow generator, flow interruption valve and a breathing circuit to which a nebuliser may be attached. IPV delivers a continuous rapid burst of gas flow to the patient’s airways via a mouthpiece or, where necessary, a mask or catheter mount. Three forms of therapy are provided during IPV: percussive oscillatory vibrations to loosen retained secretions, aerosol delivery to act as a mucolytic and positive expiratory pressure (PEP) to recruit alveolar lung units and assist in expiratory flow acceleration during a cough manoeuvre. The frequency of oscillation can be varied, as can the pressure. Short periods of breathing with IPV are interspersed with huffing and coughing to clear secretions (Newhouse et al. 1998; Pryor and Prasad 2008; Toussaint et al. 2003). Although IPV has been used in the home setting, it is better established for use in the acute setting, during admission.
Positive expiratory pressure
The application of positive expiratory pressure, applied via a facemask or mouthpiece, is believed to improve sputum clearance by its effect on peripheral airways and collateral ventilation. PEP causes an increase in lung volume, enabling air to move behind secretions, forcing them up the bronchial tree to the more central airways (Falk et al. 1984; Pryor and Prasad 2008).
The PEP mask system consists of a facemask and a one-way valve with inspiratory and expiratory ports (Figure 12.5). A resistor is attached to the expiratory port to achieve PEP. Treatment is performed in the sitting position with elbows on a table and the mask held firmly over the nose and mouth. Ten to twelve tidal breaths are taken through the mask with slight emphasis on expiration and followed by one or two huffs and coughing to clear secretions. The frequency and duration of treatment are adapted to the needs of the individual patient (average 10–20 min).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


