On completion of this chapter, the reader will be able to: • Distinguish among the various categories of anemia. • Describe the prevention of iron deficiency anemia and the care of the child with iron deficiency anemia. • Compare sickle cell anemia and β-thalassemia major in relation to pathophysiology and nursing care. • Describe the mechanisms of inheritance and nursing care of the child with hemophilia. • Relate the pathophysiology and clinical manifestations of leukemia. • Demonstrate an understanding of the rationale of therapies for neoplastic disease. • Outline a care plan for the child with neoplastic disease and the family. • Contrast the pathophysiology and management of the immunodeficiency disorders. • List nursing precautions and responsibilities during blood transfusion. • Describe the types of hematopoietic stem cell transplants. Several tests can be performed to assess hematologic function, including additional procedures to identify the cause of the dysfunction. The following discussion is limited to a description of the most common and one of the most valuable tests, the complete blood cell count (CBC). Other procedures, such as those related to iron, coagulation, and immune status, are discussed throughout the chapter as appropriate. The nurse should be familiar with the significance of the findings from the CBC (Table 43-1) and be aware of normal values for age, which are listed in Appendix B. TABLE 43-1 TESTS PERFORMED AS PART OF A COMPLETE BLOOD COUNT Average or mean quantity (weight) of Hgb in a single RBC MCH values are expressed as picograms (pg) or micromicrograms (mmcg) Whereas MCV and MCH depend on accurate counts of RBCs, MCHC does not; therefore MCHC is often more reliable All indices depend on average cell measurements and do not show individual RBC variations (anisocytosis) Index of production of mature RBCs by bone marrow Decreased count indicates depressed bone marrow function Increased count indicates erythrogenesis in response to some stimulus When reticulocyte count is extremely high, other forms of immature RBCs (normoblasts, even erythroblasts) may be present Indirectly estimates hypochromic anemia *See Appendix B for normal values according to age. Anemias are classified in relation to (1) etiology or physiology, manifested by erythrocyte or Hgb depletion, and (2) morphology, the characteristic changes in RBC size, shape, or color (Box 43-1). Although the morphologic classification is more useful in terms of laboratory evaluation of anemia, the etiologic approach provides direction for planning nursing care. For example, anemia with reduced Hgb concentration may be caused by a dietary depletion of iron and the principal intervention is replenishing iron stores. The classification of anemias is found in Fig. 43-1. In general, anemia may be suspected based on findings on the history and physical examination, such as a lack of energy, easy fatigability, and pallor, but unless the anemia is severe, the first clue to the disorder may be alterations in the CBC, such as decreased RBCs, and decreased Hgb and hematocrit (Hct) levels (see Fig. 43-1). Although anemia is sometimes defined as an Hgb level below 10 or 11 g/dL, this arbitrary cutoff is inappropriate for all children because Hgb levels normally vary with age (see Table 43-1 and Appendix B). Stool examination for occult (microscopic) blood (Hemoccult test) can identify chronic intestinal bleeding that results from a primary or secondary lactase deficiency. It is also important to understand the significance of various blood tests (see Table 43-1). Usually, several blood tests are ordered, but because they are generally done sequentially rather than at one time, the child is subjected to multiple finger or heel punctures or venipunctures. Laboratory technicians frequently are not aware of the trauma that repeated punctures represent to a child. However, these invasive procedures need not be painful (see Blood Specimens, Chapter 39). For example, the topical application of EMLA (an eutectic mix of lidocaine and prilocaine) or 4% lidocaine (Ela-Max or LMX) before needle punctures can eliminate pain (see Pain Management, Chapter 30). Therefore the nurse is responsible for preparing the child and family for the tests by: • Explaining the significance of each test, particularly why the tests are not all done at one time • Encouraging parents or another supportive person to be with the child during the procedure • Allowing the child to play with the equipment on a doll or participate in the actual procedure (e.g., by cleansing the finger with an alcohol swab) Older children may appreciate the opportunity to observe the blood cells under a microscope or in photographs. This experience is especially important if a serious blood disorder, such as leukemia, is suspected because it serves as a foundation for explaining the pathophysiology of the disorder. The following are suggested explanations for teaching children about blood components: • Red blood cells—Carry the oxygen you breathe from your lungs to all parts of your body • White blood cells—Help keep germs from causing infection • Platelets—Small parts of cells that help make bleeding stop by forming a clot (scab) over the hurt area • Plasma—The liquid portion of blood, which has clotting factors that help make bleeding stop Anemia caused by an inadequate supply of dietary iron is the most prevalent nutritional disorder in the United States and the most preventable mineral disturbance. The prevalence of iron deficiency anemia has decreased during infancy in the United States, probably in part because of families’ participation in the Women, Infants, and Children (WIC) program, which provides iron-fortified formula for the first year of life and routine screening of Hgb levels during early childhood (Baker, Greer, and Committee on Nutrition American Academy of Pediatrics [AAP], 2010; Cusick, Mei, Freedman, et al., 2008). Preterm infants are especially at risk because of their reduced fetal iron supply. Children 12 to 36 months of age are at risk for anemia as a result of primarily cow’s milk intake and not eating an adequate amount of iron-containing food (Andrews, Ullrich, and Fleming, 2009; Baker, Greer, and Committee on Nutrition AAP, 2010; Richardson, 2007). Adolescents are also at risk because of their rapid growth rate combined with poor eating habits, menses, obesity, or strenuous activities. In formula-fed infants, the most convenient and best sources of supplemental iron are iron-fortified commercial formula and iron-fortified infant cereal. Iron-fortified formula provides a relatively constant and predictable amount of iron and is not associated with an increased incidence of gastrointestinal (GI) symptoms, such as colic, diarrhea, or constipation. Infants younger than 12 months should not be given fresh cow’s milk because it may increase the risk for GI blood loss occurring from exposure to a heat-labile protein in cow’s milk or cow’s milk–induced GI mucosal damage resulting from a lack of cytochrome iron (heme protein) (Glader, 2007; Richardson, 2007). If GI bleeding is suspected, the child’s stool should be guaiac tested on at least four or five occasions to identify any intermittent blood loss. If the Hgb level fails to rise after 1 month of oral therapy, it is important to assess for persistent bleeding, iron malabsorption, noncompliance, improper iron administration, or other causes of the anemia. Parenteral (IV or intramuscular [IM]) iron administration is safe and effective but painful, expensive, and occasionally associated with regional lymphadenopathy, transient arthralgias, or serious allergic reaction (Andrews, Ullrich, and Fleming, 2009; Glader, 2007; McKenzie, 2004). Therefore parenteral iron is reserved for children who have iron malabsorption or chronic hemoglobinuria. Transfusions are indicated for the most severe anemia and in cases of serious infection, cardiac dysfunction, or surgical emergency when anesthesia is required. Packed RBCs (2-3 mL/kg), not whole blood, are used to minimize the chance of circulatory overload. Supplemental oxygen is administered when tissue hypoxia is severe. The prognosis for a child with this condition is very good. However, some evidence indicates that if the iron deficiency anemia is severe and longstanding, cognitive, behavioral, and motor impairment may result (Andrews, Ullrich, and Fleming, 2009; Lokeshwar, Mehta, Mehta, et al., 2011; McCann and Ames, 2007). Cow’s milk contains substances that bind the iron and interfere with absorption. Iron supplements should not be administered with milk or milk products (Carley, 2003). If parenteral iron preparations are prescribed, iron dextran must be injected deeply into a large muscle mass using the Z-track method. The injection site is not massaged after injection to minimize skin staining and irritation. Because no more than 1 mL should be given in one site, the IV route should be considered to avoid multiple injections. Careful observation is required because of the risk for adverse reactions, such as anaphylaxis, with IV administration. A test dose is recommended before routine use. Recently, a new IV iron preparation (ferumoxytol) was approved in the United States that shows promise in complete replacement of iron with little toxicity (Auerbach, 2011). A primary nursing objective is to prevent nutritional anemia through family education. Because breast milk is a low iron source, the nurse must reinforce the importance of administering iron supplementation to exclusively breastfed infants by 4 months of age (Baker, Greer, and Committee on Nutrition AAP, 2010; Lokeshwar, Mehta, Mehta, et al., 2011). The American Academy of Pediatrics (AAP) recommends preterm, marginally low–birth-weight and low-birth-weight infants or infants with inadequate iron stores at birth receive iron supplements at approximately 2 months of age (Berglund, Westrup, and Domellof, 2010). In formula-fed infants, the nurse discusses with parents the importance of using iron-fortified formula and introducing solid foods at the appropriate age during the first year of life. Traditionally, cereals are one of the first semisolid foods to be introduced into the infant’s diet at approximately 6 months of age (Baker, Greer, and Committee on Nutrition AAP, 2010; Glader, 2007; Lokeshwar, Mehta, Mehta, et al., 2011). The best solid-food source of iron is commercial iron-fortified cereals. It may be difficult at first to teach the infant to accept foods other than milk. The same principles are applied as those for introducing new foods (see Nutrition, Chapter 31), especially feeding the solid food before the milk. Predominantly milk-fed infants rebel against solid foods, and parents are cautioned about this and the need to be firm in not relinquishing control to the child. It may require intense problem solving on the part of both the family and the nurse to overcome the child’s resistance. The following are the most common forms of SCD in the United States: • SCA, the homozygous form of the disease (HbSS or SS) • Sickle cell–C disease, a heterozygous variant of SCD, including both HbS and HbC (SC) • Sickle cell–hemoglobin E disease, a variant of SCD in which glutamic acid has been substituted for lysine in the number 26 position of the β-chain (SE) • Sickle thalassemia disease, a combination of sickle cell trait and β-thalassemia trait (Sβthal). β+ refers to the ability to still produce some normal HbA. β0 indicates that there is no ability to produce HbA. Of the SCDs, SCA is the most common form in African-Americans, followed by sickle cell–C disease and sickle thalassemia. Sickle syndromes exist when the HbS is paired with other mutant globins. Sickle cell disease is one of the most common genetic diseases worldwide. SCD affects approximately 90,000 Americans, primarily African-American, followed by Hispanics, with a lower incidence in the other ethnic groups (Driscoll, 2007). The incidence of the disease varies in different geographic locations. Among African-Americans, the incidence of sickle cell trait is about 9%. In West Africa, the incidence is reported to be as high as 40% among native Africans. The high incidence of sickle cell trait in West Africans is believed by some to be the result of selective protection afforded trait carriers against one type of malaria. Although the defect is inherited, the sickling phenomenon is usually not apparent until later in infancy because of the presence of fetal Hbg (HbF). As long as the child has predominantly HbF, sickling does not occur because there is less HbS. Newborns with SCA are generally asymptomatic because of the protective effect of HbF (60%-80% HbF), but this rapidly decreases during the first year, so these children are at risk for sickle cell–related complications (Driscoll, 2007; Heeney and Dover, 2009). The clinical features of SCA are primarily the result of (1) obstruction caused by the sickled RBCs, (2) vascular inflammation, and (3) increased RBC destruction (Fig. 43-2). The abnormal adhesion, entanglement, and enmeshing of rigid sickle-shaped cells accompanied by the inflammatory process intermittently block the microcirculation, causing vasoocclusion (Fig. 43-3). The resultant absence of blood flow to adjacent tissues causes local hypoxia, leading to tissue ischemia and infarction (cellular death). Most of the complications seen in SCA can be traced to this process and its impact on various organs of the body (Box 43-2). Another serious complication is acute chest syndrome (ACS), which is clinically similar to pneumonia. It is the presence of a new pulmonary infiltrate and may be associated with chest pain, fever, cough, tachypnea, wheezing, and hypoxia. A cerebrovascular accident (CVA, stroke) is a sudden and severe complication, often with no related illnesses. Sickled cells block the major blood vessels in the brain, resulting in cerebral infarction, which causes variable degrees of neurologic impairment. The current treatment for SCD children who have experienced a stroke is chronic transfusion therapy. Repeat CVAs causing progressively greater brain damage occur in approximately 70% of untreated children who have experienced one stroke (Heeney and Dover, 2009). Administration of pneumococcal and meningococcal vaccines is recommended for these children because of their susceptibility to infection as a result of functional asplenia. In addition to routine immunizations, children with SCD should receive a yearly influenza vaccination (see Immunizations, Chapter 31). Oral penicillin prophylaxis is also recommended by 2 months of age to reduce the chance of pneumococcal sepsis (see Evidence-Based Practice box) (AAP Committee on Infectious Diseases, 2009; Hirst and Owusu-Ofori, 2010; National Institutes of Health, 2002; Pack-Mabien and Haynes, 2009). Oxygen therapy is of little therapeutic value unless the patient has hypoxia (Heeney and Dover, 2009). Severe hypoxia must be prevented because it causes massive systemic sickling that can be fatal. Oxygen administration is usually not effective in reversing sickling or reducing pain because the oxygen is unable to reach the enmeshed sickled erythrocytes in clogged vessels (Chiocca, 1996). In addition, prolonged administration of oxygen can depress bone marrow, further aggravating the anemia (Khoury and Grimsley, 1995). Another important component of care is the use of blood transfusions. Exchange RBC transfusion (erythrocytapheresis) is the replacement of sickle cells with normal RBCs. Exchange transfusion is a successful, rapid method of reducing the number of circulating sickle cells and therefore slowing down the vicious circle of hypoxia, thrombosis, tissue ischemia, and injury. The procedure is advocated as a possible technique in preventing reoccurrence of ACS and CVA (Velasquez, Mariscalco, Goldstein, et al., 2009). A transcranial Doppler (TCD) test identifies the child with SCD who is at high risk for developing a CVA by monitoring the intracranial vascular flow (Driscoll, 2007; Kwiatkowski, Yim, Miller, et al., 2011). The TCD test is performed yearly for children from 2 to 16 years of age. The recommended treatment for children with confirmed abnormal TCD is chronic transfusion therapy (Armstrong-Wells, Grimes, Sidney, et al., 2009; Driscoll, 2007; Kwiatkowski, Yim, Miller, et al., 2011). Multiple transfusions carry the risk for transmission of viral infection, hyperviscosity, transfusion reactions, alloimmunization, and hemosiderosis (Driscoll, 2007; Heeney and Dover, 2009). After a CVA, blood transfusions are usually given every 3 to 4 weeks to help prevent a repeat stroke. To reduce iron overload from chronic transfusion therapy, chelation therapy may be started (see p. 1311). The most common and debilitating symptom experienced by patients with SCD is VOC, which is accompanied by increasing heath care cost because of prolonged hospitalization associated with pulmonary and GI complications (Driscoll, 2007; Raphael, Mei, Mueller, et al., 2012). The chronic nature of this pain can greatly affect the child’s development. A multidisciplinary team (e.g., physician, psychologist, family, nurse, social worker) approach is best for vasoocclusive pain management that includes pharmacologic treatment, hydration, physical therapy, and complementary treatment (e.g., prayer, spiritual healing, massage, herbs, relaxation, acupuncture, and biofeedback) (Brandow, Weisman, and Panepinto, 2011; Redding-Lallinger and Knoll, 2006). When mild to moderate VOC is reported, nonsteroidal antiinflammatory medication (e.g., ibuprofen, ketorolac) or acetaminophen (Tylenol) is used initially. If these drugs are not effective alone, codeine can be added. The dosages of both drugs are titrated (adjusted) to a therapeutic level. Opioids such as immediate- and sustained-release morphine, oxycodone, hydromorphone (Dilaudid), and methadone are administered intravenously or orally for severe pain and are given around the clock. In conjunction with the opioid, IV ketorolac for a maximum of a 5-day course is commonly used to enhance the pain management effect. Patient-controlled analgesia (PCA) has been used successfully for sickle cell–related pain. PCA reinforces the patient’s role and responsibility in managing the pain and provides flexibility in dealing with pain, which may vary in severity over time (see Pain Management, Chapter 30). Meperidine (Demerol) is not recommended. Normeperidine, a metabolite of meperidine, is a central nervous system (CNS) stimulant that produces anxiety, tremors, myoclonus, and generalized seizures when it accumulates with repetitive dosing. Patients with SCD are particularly at risk for normeperidine-induced seizures (Howard and Davies, 2007; National Institutes of Health, 2002). The prognosis varies, but most patients live into the fifth decade. Most of the time, children are without symptoms and participate in normal activities without restrictions. The greatest risk is usually in children younger than 5 years, and the majority of deaths in these children are caused by overwhelming infection. Consequently, SCA is a chronic illness with a potentially terminal outcome. Physical and sexual maturation are delayed in adolescents with SCA. Although adults achieve normal height, weight, and sexual function, the delay may present problems to adolescents (Heeney and Dover, 2009; Redding-Lallinger and Knoll, 2006). Individuals with SCD who have higher levels of HbF tend to have a milder disease with fewer complications than those with lower levels (Anderson, 2006; Driscoll, 2007). Hydroxyurea is a U.S. Food and Drug Administration–approved medication that increases the production of HbF, reduces endothelial adhesion of sickle cells, improves the sickle cell hydration, increases nitric oxide production (a vasodilator), and lowers leukocyte and reticulocyte counts (McGann and Ware, 2011; National Institutes of Health, 2002). Long-term follow-up of patients taking hydroxyurea alone revealed a 40% reduction in mortality and decreased frequency of VOC, ACS, hospital admissions, and need for transfusions, thus making SCD crises milder (Anderson, 2006; Strouse, Lanzkron, Beach, et al., 2008). Pediatric studies have shown that hydroxyurea can be safely used in children (Wang, Ware, Miller, et al., 2011; Zimmerman, Schultz, Davis, et al., 2004). Hematopoietic stem cell transplantation (HSCT) offers a curative approach for some children with SCD with event-free survival of 95% (Driscoll, 2007; Haining, Duncan, and Lehmann, 2009) (see p. 1389). The success of many of the medical therapies relies heavily on nursing implementation. Management of pain is an especially difficult problem and often involves experimenting with various analgesics, including opioids, and schedules before relief is achieved. Unfortunately, these children tend to be undermedicated, resulting in their “clock watching” and demands for additional doses sooner than might be expected. Often this incorrectly raises suspicions of drug addiction, when in fact the problem is one of improper dosage (see Family-Centered Care box). In choosing and scheduling analgesics, the goal should be prevention of pain. Fear of Addiction Although the pain during a sickle cell crisis is usually severe and opioids are needed, many families fear that their child will become addicted to the narcotic. Unfortunately, misinformed health care professionals may foster this unfounded fear, which results in needless suffering. Extremely few children who receive opioids for severe pain become behaviorally addicted to the drug (American Pain Society, 1999; Howard and Davies, 2007; National Institutes of Health, 2002). Families and older children, especially adolescents, need to be reassured that opioids are medically indicated, high doses may be needed, and children rarely become addicted. In splenic sequestration, the size of the spleen is gently measured by abdominal palpation (see Abdomen, Chapter 29). The nurse should be aware of spleen size because increasing splenomegaly is an ominous sign. A decreasing spleen size denotes response to therapy. Vital signs and blood pressure are also closely monitored for impending shock. Anemia is typically not a presenting complication in vasoocclusive crises but is a critical problem in other types of crises. The nurse monitors for evidence of increasing anemia and institutes appropriate nursing interventions. Oxygen is not beneficial in vasoocclusive episodes unless hypoxemia is present (Heeney and Dover, 2009). It does not reverse sickled RBCs, and if used in a nonhypoxic patient, it will decrease erythropoiesis (Vichinsky and Styles, 1996). Because prolonged use of oxygen can aggravate the anemia, signs of lack of therapeutic benefit, such as restlessness, increased pallor, and continued pain, are reported. Families need the opportunity to discuss their feelings regarding transmitting a potentially fatal, chronic illness to their child. Because of the widely publicized prognosis for children with SCA, many parents express their prevalent fear of the child’s death. Three manifestations of SCD that may appear in the first 2 years of life (dactylitis, severe anemia, leukocytosis) can be predictors of disease severity (DeBaun and Vichinsky, 2007; Ohls and Christensen, 2007). The nurse should care for the family as for any family with a child who has a chronic and life-threatening illness and consider the siblings’ reactions, the stress on the marital relationship, and the childrearing attitudes displayed toward the child. Several resources are available to families with a sickling disorder.* Worldwide, thalassemia is a common genetic disorder, affecting as many as 15 million people (Yaish, 2010). The term thalassemia, which is derived from the Greek word thalassa, meaning “sea,” is applied to a variety of inherited blood disorders characterized by deficiencies in the rate of production of specific globin chains in Hgb. The name appropriately refers to descendants of or people living near the Mediterranean Sea, who have the highest incidence of the disease, namely Italians, Greeks, and Syrians. Evidence suggests that the high incidence of the disorders among these groups is a result of the selective advantage the trait confers in relation to malaria, as is postulated in SCD. However, the disorder has a wide geographic distribution, probably as a result of genetic migration through intermarriage or possibly as a result of spontaneous mutation. β-Thalassemia is the most common of the thalassemias and occurs in four forms: • Two heterozygous forms, thalassemia minor, an asymptomatic silent carrier; and thalassemia trait, which produces a mild microcytic anemia • Thalassemia intermedia, which is manifested as splenomegaly and moderate to severe anemia • A homozygous form, thalassemia major (also known as Cooley anemia), which results in a severe anemia that would lead to cardiac failure and death in early childhood without transfusion support The onset of thalassemia major may be insidious and not recognized until the latter half of infancy. The clinical effects of thalassemia major are primarily attributable to (1) defective synthesis of HbA, (2) structurally impaired RBCs, and (3) shortened life span of erythrocytes (Box 43-3).
Hematologic and Immunologic Dysfunction
Hematologic Dysfunction
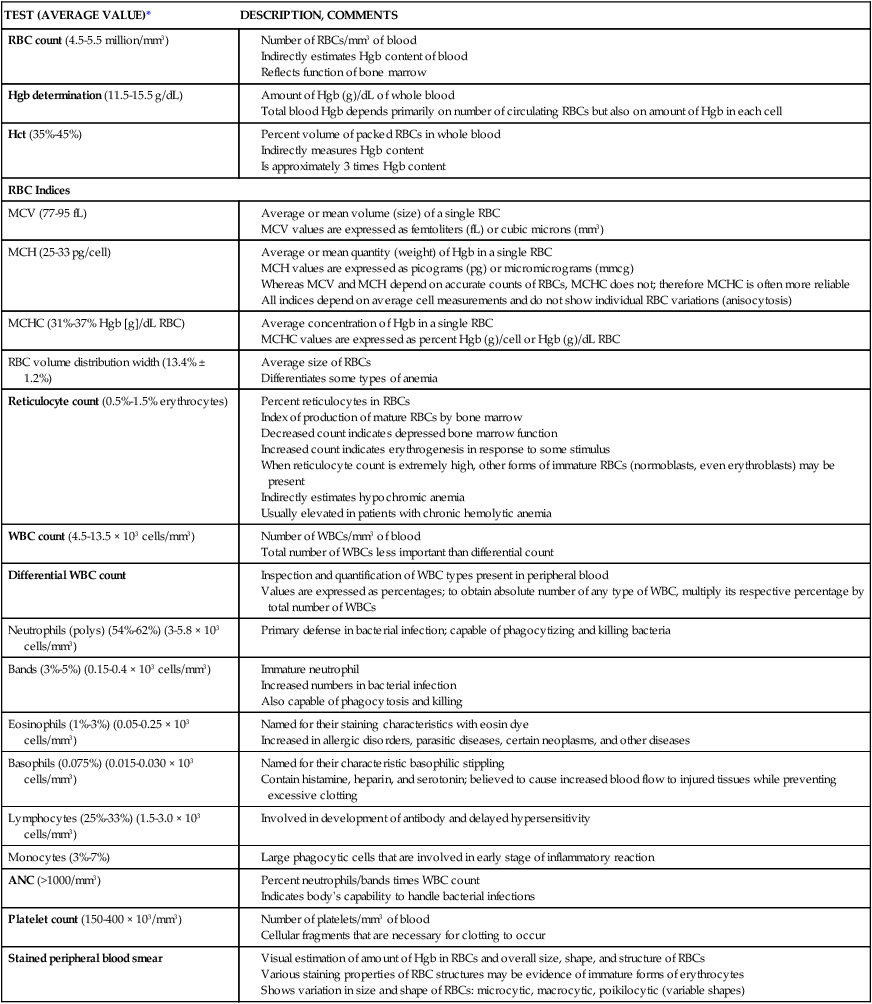
TEST (AVERAGE VALUE)*
DESCRIPTION, COMMENTS
RBC count (4.5-5.5 million/mm3)
Hgb determination (11.5-15.5 g/dL)
Hct (35%-45%)
RBC Indices
MCV (77-95 fL)
MCH (25-33 pg/cell)
MCHC (31%-37% Hgb [g]/dL RBC)
RBC volume distribution width (13.4% ± 1.2%)
Reticulocyte count (0.5%-1.5% erythrocytes)
WBC count (4.5-13.5 × 103 cells/mm3)
Differential WBC count
Neutrophils (polys) (54%-62%) (3-5.8 × 103 cells/mm3)
Bands (3%-5%) (0.15-0.4 × 103 cells/mm3)
Eosinophils (1%-3%) (0.05-0.25 × 103 cells/mm3)
Basophils (0.075%) (0.015-0.030 × 103 cells/mm3)
Lymphocytes (25%-33%) (1.5-3.0 × 103 cells/mm3)
Monocytes (3%-7%)
ANC (>1000/mm3)
Platelet count (150-400 × 103/mm3)
Stained peripheral blood smear
Red Blood Cell Disorders
Anemia
Classification
Diagnostic Evaluation
Care Management
Prepare the Child and Family for Laboratory Tests.
 Nursing Alert
Nursing Alert
Iron Deficiency Anemia
Therapeutic Management
Prognosis.
Care Management
 Medication Alert
Medication Alert
Diet.
Sickle Cell Anemia
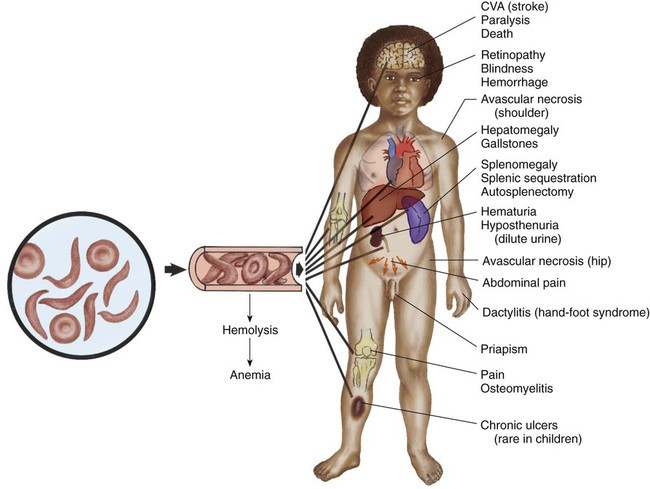
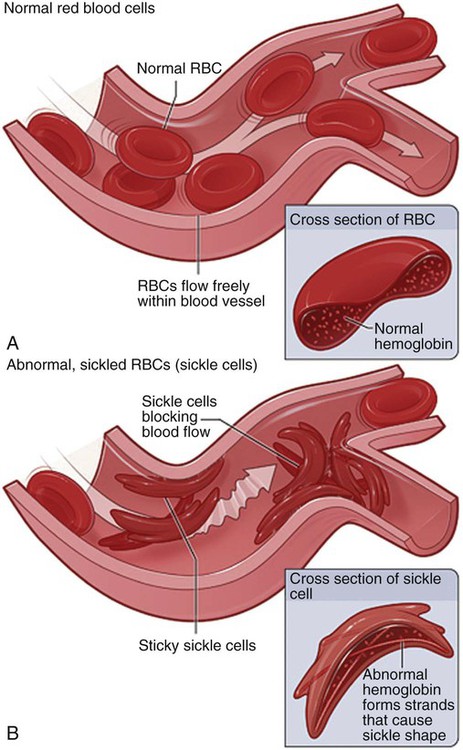
Pathophysiology
Therapeutic Management
 Medication Alert
Medication Alert
Prognosis.
Care Management
Promote Supportive Therapies During Crises.
 Family-Centered Care
Family-Centered Care
Support the Family.
β-Thalassemia (Cooley Anemia)
Diagnostic Evaluation
 Nursing Alert
Nursing Alert
 Nursing Alert
Nursing Alert Medication Alert
Medication Alert Nursing Alert
Nursing Alert Nursing Alert
Nursing Alert


