Human genetics, as it pertains to health care, is the study of the etiology, pathogenesis, and natural history of human conditions that are influenced by genetic factors. Genetic factors extend beyond the limited view of solely distinct genetic syndromes to encompass influences on health, the occurrence of complex disorders, individual biologic responses to illness, potential treatment and medical management approaches, and strategies for prevention or cure.
This tremendous realization is apparent through the accomplishments of the Human Genome Project. This 15-year international collaborative effort was completed in 2003. One significant goal of the Human Genome Project was to identify the approximately 25,000 human genes. These advances and the associated knowledge will continue to affect the delivery of health care and nursing practice significantly. Genetic evaluations, screening, testing, guided treatment, family counseling, and related legal, ethical, and psychosocial issues are becoming daily practice for many nurses.
The impact of genetics on nursing is significant. In 1997, the American Nurses Association (ANA) officially recognized genetics as a nursing specialty. This effort was spearheaded by the International Society of Nurses in Genetics (ISONG), which also initiated credentialing for the Advanced Practice Nurse in Genetics and the Genetics Clinical Nurse. ANA and ISONG have collaborated in the establishment of a scope and standards of practice for nurses in genetics practice. Essential Nursing Competencies and Curricula Guidelines for Genetics and Genomics were finalized in 2006 with Outcome Indicators established in 2008. They reflect the minimal genetic and genomic competencies for every nurse regardless of academic preparation, practice setting, role, or specialty. A copy of these competencies is available through the ANA or the National Human Genome Research Institute websites (www.genome.gov/17517146).
The purpose of this chapter is to provide the nurse with practical information, resources, representative examples, and professional considerations critical to integration of genetics knowledge into nursing practice.
Underlying Principles
Readers are encouraged to use the Talking Glossary of Genetic Terms available through the National Human Genome Research Institute to supplement the terms provided here. This glossary can be found at www.genome.gov/glossary.
Cell: The Basic Unit of Biology
Cytoplasm—contains functional structures important to cellular functioning, including mitochondria, which contain extranuclear deoxyribonucleic acid (DNA) important to mitochondrial functioning.
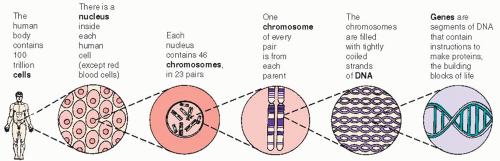
Nucleus—contains 46 chromosomes in each somatic (body) cell, or 23 chromosomes in each germ cell (egg or sperm) (see Figure 4-1, page 34).
Chromosomes
Each somatic cell with a nucleus has 22 pairs of autosomes (the same in both sexes) and 1 pair of sex chromosomes.
Females have two X sex chromosomes; males have one Y sex chromosome and one X sex chromosome.
Normally, at conception, each individual receives one copy of each chromosome from the maternal egg cell (1 genome) and one copy of each chromosome from the paternal sperm cell (1 genome), for a total of 46 chromosomes (2 genomes).
Karyotype is the term used to define the chromosomal complement of an individual (eg, 46, XY), as is determined by laboratory chromosome analysis.
Each chromosome contains a different number of genes, ranging from approximately 380 to 3,000 genes.
Figure 4-1. Cells, chromosomes, DNA, and genes.
Genes
The basic unit of inherited information.
Each copy of the human genome in the nucleus has about 25,000 genes. Cells also have some non-nuclear genes located within the mitochondria within the cytoplasm.
Alternate forms of a gene are termed alleles.
For each gene, an individual receives one allele from each parent, and thus has two alleles for each gene on the autosomes and also on the X chromosomes in females.
Males have only one X chromosome and, therefore, have only one allele for all genes on the X chromosome; they are hemizygous for all X-linked genes.
At any autosomal locus, or gene site, an individual can have two identical alleles (homozygous) for that locus or can have two different alleles (heterozygous) at a particular locus.
Genotype refers to the constitution of the genetic material of an individual; for practical purposes, it is commonly used to refer to a particular base or bases in the DNA. For example, the gene for sickle cell disease, the gene for cystic fibrosis, or the gene for familial polyposis.
Phenotype refers to the physical or biochemical characteristics an individual manifests regarding expression of the presence of a particular feature, or set of features, associated with a particular gene.
Each gene is composed of a unique sequence of DNA bases.
DNA: Nuclear and Mitochondrial
Human DNA is a double-stranded helical structure comprised of four different bases, the sequence of which codes for the assembly of amino acids to make a protein—for example, an enzyme. These proteins are important for the following reasons:
For body characteristics such as eye color.
For biochemical processes such as the gene for the enzyme that digests phenylalanine.
For body structure such as a collagen gene important to connective tissue and bone formation.
For cellular functioning such as genes associated with the cell cycle.
The four DNA bases are adenine, guanine, cytosine, and thymine (A, G, C, and T).
A change, or mutation, in the coding sequence, such as a duplicated or deleted region, or even a change in only one base, can alter the production or functioning of the gene or gene product, thus affecting cellular processes, growth, and development.
DNA analysis can be done on almost any body tissue (blood, muscle, skin) using molecular techniques (not visible under a microscope) for mutation analysis of a specific gene with a known sequence or for DNA linkage of genetic markers associated with a particular gene.
Normal Cell Division
Mitosis occurs in all somatic cells, which, under normal circumstances, results in the formation of cells identical to the original cell with the same 46 chromosomes.
Meiosis, or reduction division, occurs in the germ cell line, resulting in gametes (egg and sperm cells) with only 23 chromosomes, one representative of each chromosome pair.
During the process of meiosis, parental homologous chromosomes (from the same pair) pair and undergo exchanges of genetic material, resulting in recombinations of alleles on a chromosome and thus variation in individuals from generation to generation.
CLINICAL APPLICATION
Genetic Disorders
Presentations warranting genetic consideration include mental retardation, birth defects, biochemical or metabolic disorders, structural abnormalities, multiple miscarriages, and family history of the same or related disorder.
Examples of disorders that result from abnormalities of chromosomes or genes or that are, at least in part, influenced by genetic factors are described in Table 4-1.
Classification of Genetic Alterations
Chromosomal
The entire chromosome or only part can be affected. This is usually associated with birth defects and mental retardation because there are extra or missing copies of all genes associated with the involved chromosome.
Numerical—abnormal number of chromosomes due to nondisjunction (error in chromosomal separation during cell division). Examples are Down and Klinefelter syndromes.
Structural—abnormality involving deletions, additions, or translocations (rearrangements) of parts of chromosomes. Examples are Prader-Willi and Angelman syndromes.
Fragile sites—regions susceptible to chromosomal breakage such as in fragile X syndrome.
May involve autosomes or sex chromosomes.
Table 4-1 Selected Genetic Disorders
DISORDER AND INCIDENCE
CHARACTERISTICS
ETIOLOGY AND RECURRENCE RISKS
CONSIDERATIONS AND COMMENTS
Chromosomal Disorders
Autosomal
Down syndrome (Trisomy 21) 1 in 700 neonates; incidence increases with advanced maternal age (eg, risk at maternal age 25 is 1 in 1,350; at age 35, 1 in 384; at age 45, 1 in 28)
• Extra copy of number 21 chromosome (total of three copies).
• 94% of cases are trisomy (karyotype 47, +21) for three distinct number 21 chromosomes due to nondisjunction (failure of chromosomal separation during meiosis); recurrence risk 1%, plus maternal age-related risk if older than age 35.
• 4% of cases have a translocation—the extra number 21 is attached to another chromosome, usually a number 13 or number 14; half of these translocations are new occurrences, the other half are inherited from a parent.
• 2% of cases are mosaic— affected individual has two different cell lines, one with the normal number of chromosomes and the other cell line trisomic for the number 21 chromosome; due to a postconception error in chromosomal division during mitosis.
• Recurrence risk for parents of affected are dependent on one or more of the following: chromosomal type of disorder, maternalage, parental karyotype, family history, and sex of transmitting parent and other chromosome involved (if translocation).
• May demonstrate nuccal thickening prenatally on ultrasound examination.
• Associated with moderate mental retardation.
• No phenotypic differences between trisomy Down syndrome and translocation Down syndrome.
• Chromosome analysis should be performed on all persons with Down syndrome.
• Prenatal maternal serum screening can adjust risk for the pregnancy.
Trisomy 13 (Patau syndrome) 1 in 5,000 live births
Holoprosencephaly; cleft lip or palate, or both; abnormal helices; cardiac defects; rocker-bottom feet; overlapping positioning of fingers; seizures; severe mental retardation
• Extra number 13 chromosome (total of three copies): Either trisomy form, due to nondisjunction, with less than a 1% recurrence risk; or translocation form, with recurrence risk less than that of translocation Down syndrome and dependent on other factors, including chromosomes involved.
• 44% die within the first month; 18% survive first year of life.
Trisomy 18 (Edwards syndrome) 1 in 6,000 live births
Small for gestational age (may be detected prenatally); feeble fetal activity; weak cry; prominent occiput; low-set, malformed ears; short palpebral fissures; small oral opening; overlapping positioning of fingers (fifth digit over fourth, index over third); nail hypoplasia, short hallux; cardiac defects; inguinal or umbilical hernia; cryptorchidism in males; severe mental retardation
• Extra number 18 chromosome (total of three copies): Majority due to trisomy with less than 1% recurrence risk.
• Most trisomy 18 conceptions miscarry; 90% die within first year of life.
Sex Chromosome
Klinefelter syndrome 1 in 700 males; 47, XXY abnormality in 90%; other 10% have more than two X chromosomes in addition to the Y chromosome or have mosaicism (about 20%)
Body habitus may be tall, slim, and underweight; long limbs; gynecomastia; small testes; inadequate virilization; azoospermia or low sperm count; cognitive defects; behavioral problems
• Due to nondisjunction during meiosis, except for cases of mosaicism, which are due to mitotic nondisjunction.
• No distinguishing features prenatally.
• Diagnosis may not be suspected or pursued before puberty.
• Diagnosis in childhood is beneficial in planning for testosterone replacement therapy, in addition to accurate understanding of learning or behavioral problems.
• Tend to be delayed in onset of speech, have difficulty in expressive language; may be relatively immature; may have history of recurrent respiratory infections.
Turner syndrome (45, X) 1 in 2,500 female births
Webbing of neck and short stature; lymphedema of hands and feet as neonate; congenital cardiac defects (especially coarctation of the aorta); low posterior hairline; cubitus valgus; widely spaced nipples; underdeveloped breasts; immature internal genitalia (eg, streak ovaries); primary amenorrhea; learning disabilities
• About 50% due to a nondisjunctional error during meiosis (karyotype 45, X); 20% are mosaic due to nondisjunction during mitosis; 30% have two X chromosomes but one is functionally inadequate (eg, due to presence of abnormal gene); generally a sporadic occurrence.
• Webbing of neck and short stature may be detected prenatally by ultrasound.
• Early diagnosis enhances optimal health care management (eg, planning for administration of growth hormone therapy, estrogen replacement).
• Psychosocial implications associated with short stature, delayed onset of puberty.
• Infertility associated with ovarian dysgenesis; oocyte donation and adoption are generally the only options for having children.
Microdeletion/Microduplication
Fragile X 1 in 1,200 males; 1 in 2,500 females
Motor delays; hypotonia; speech delay and language difficulty; hyperactivity; classic features including long face, prominent ears, and macroorchidism manifest around puberty; autism (about 7% of males); mental retardation in most males; learning disabilities in most affected females
• Mutation in the fragile X menta l retardation gene (FMR-1), represented as a large DNA expansion of a normally present trinucleotide.
• Carrier mother of an affected male has a 50% risk for future affected males and 50% chance of transmitting the FMR-1 X chromosome to a daughter who would be a carrier, may be unaffected, or manifest features associated with the fragile X syndrome and has a 50% chance of transmitting that gene to future offspring.
Only gold members can continue reading. Log In or Register to continue