14 Gastrointestinal and Nutritional Disorders
 Be sure to check out the supplementary content available at
Be sure to check out the supplementary content available at Pearls
• Abdominal distension can compromise diaphragm expansion and can lead to respiratory insufficiency.
• A child with significant intraabdominal pathology may have subtle clinical signs, such as somnolence and mental status changes.
• Bilious vomiting with a clinical examination suggestive of an acute abdomen can be a sign of a bowel-threatening pathologic condition (e.g., volvulus) and almost always warrants immediate operative intervention.
• If a child does not recover as expected after gastrointestinal surgery, a technical complication of the procedure should be considered as a cause until it is ruled out.
• Correct placement of a nasogastric or orogastric tube is verified with an abdominal radiograph.
• Enteral feedings should begin as soon as possible, because even small quantities of feedings reduce the risk of infection and promote gut healing.
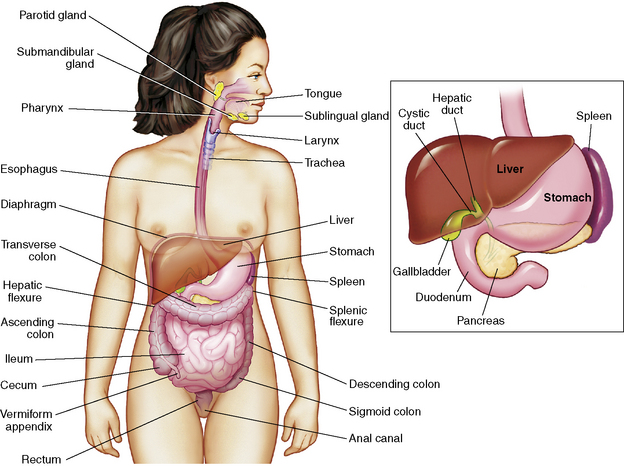
Essential anatomy and physiology
This section summarizes the basic anatomy and physiology of major structures composing the GI tract (Fig. 14-1). See the Chapter 14 Supplement on the Evolve Website for additional information on maturational anatomy and physiology.
Stomach
Secreted mucus forms a protective barrier between the mucosa and the acid and proteolytic enzymes. Acid produced by partial cells creates a gastric pH of 1 to 2 that dissolves food fiber, acts as a bactericide against swallowed organisms, and converts pepsinogen to pepsin. Pepsinogen arises from chief cells. Under the influence of gastric acid, pepsinogen is converted into pepsin, a proteolytic enzyme that continues the breakdown of proteins that was started by gastric acids. Intrinsic factor is a glycoprotein required for vitamin B12 absorption.27
Small Intestine
The small intestine (Fig. 14-2) begins beyond the pylorus of the stomach and is divided into the duodenum (receives enzymes important for digestion), the jejunum (principle absorbing site), and the ileum (the only site for the absorption of vitamin B12 and bile acids). Two layers of smooth muscle, an outer longitudinal layer, and an inner thicker circular layer produce peristalsis.
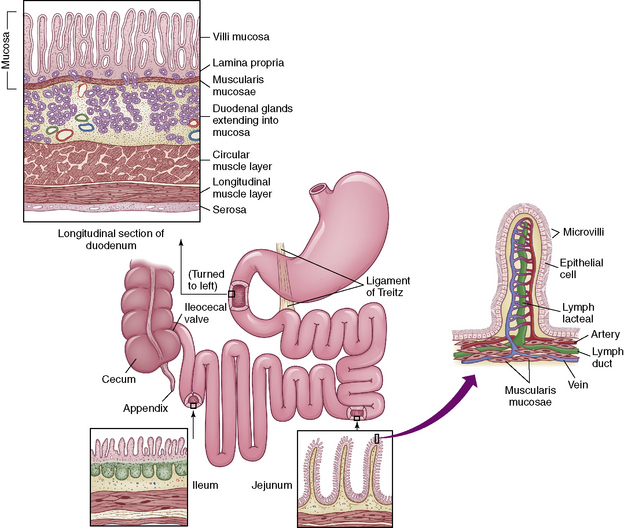
Fig. 14-2 The small intestine.
(From McCance KL, Heuther SE, editors: Pathophysiology: the biologic basis for disease in adults and children, ed 6, St Louis, 2010, Elsevier.)
Liver
The liver is one of the largest organs of the body, and it performs hundreds of functions. It is divided into right and left lobes by the falciform ligament (Fig. 14-3). The right lobe is the largest lobe, composed of the right lobe proper, the caudate lobe (posterior surface), and the quadrate lobe (inferior surface).
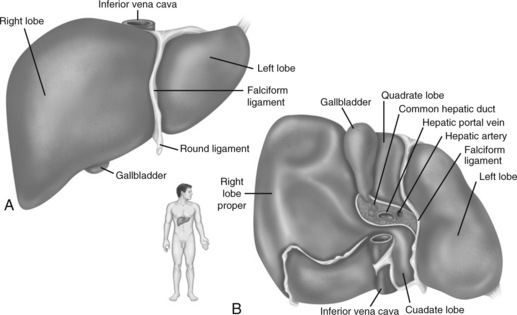
Fig. 14-3 Gross structure of the liver. A, Anterior view. B, Inferior view.
(From Patton KT, Thibodeau GA: Anatomy and physiology, ed 7, St Louis, 2010, Mosby.)
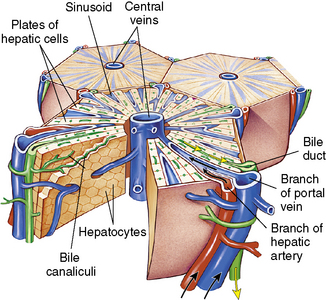
The liver has 50,000-100,000 functional units or lobules composed of hepatic plates (i.e., plates of hepatocytes) that each radiate centrally around a central vein (Fig. 14-4). The porta hepatis is a fissure that serves as the entry point for the hepatic artery, the portal vein, and the common bile duct. The artery, vein, and duct divide into intralobular branches as they follow the septa throughout the liver.
Pancreas
The pancreas is composed of a head, a body, and a tail. The head is tucked into the curve of the duodenum, and the tail reaches laterally across the midline to the spleen. The pancreas is a retroperitoneal organ and is well protected from most environmental impacts. It functions as both an exocrine and endocrine organ. The working cells of the exocrine pancreas are acini cells; these cells are organized as spheres encompassing small secretory ducts scattered throughout the organ. This series of ducts transports fluid containing the digestive enzymes (proteases, amylase, and lipase) and sodium bicarbonate (pH 7.1-8.2) produced by acini cells to the pancreatic duct.27 The pancreatic duct runs the length of the organ and then drains into the common bile duct at the ampulla of Vater, ultimately draining into the duodenum through the sphincter of Oddi.
Nutrition
There is little evidence to identify the best nutritional support of the critically ill child.30,44 A recent Cochrane review that attempted to assess the impact of enteral and parenteral nutrition (PN) on clinically important outcomes in the critically ill child found only one trial that was relevant to the review question.30 Appropriate nutritional assessment, determination of energy requirements, and the timing, route, and type of nutritional delivery have yet to be established for seriously ill or injured children in the critical care unit.
Recommended Daily Nutritional Intake
Adequate energy (caloric) intake is necessary for rapid growth and development throughout childhood. During infancy the requirement for energy intake per kilogram is greater than in all other age groups. The average requirements for energy intake during infancy and childhood are shown in Table 14-1. Of note, these requirements are for healthy, active infants and children.
Table 14-1 Daily Energy Requirements for Infants and Children
| Age | kcal/kg |
| Up to 6 months | 90-110 |
| 6-12 months | 80-100 |
| 12-36 months | 75-90 |
| 4-6 years | 65-75 |
| 7-10 years | 55-75 |
| 11-18 years | 40-55 |
It is useful to divide energy requirements into resting energy expenditure (REE) and requirements for growth and physical activity. REE is determined by the basal metabolic rate or the energy consumed for the normal maintenance of cellular energy. Estimates of an ill child’s REE by standard equations are unreliable, and indirect calorimetry is not always available to clinicians.44 Caloric requirements for the hospitalized child may be substantially more or less than the normal daily recommended requirements. Hypermetabolic states, excessive nutrient losses, trauma, burns, and surgery will increase energy requirements, whereas neurologic impairment, reduced activity, and ventilator support can decrease energy needs. Typically, fever increases energy requirements 12% per day for each degree Celsius elevation in temperature above 37° C.
Fluid and electrolyte requirements vary as a function of age, weight, and clinical condition. Normal daily fluid and electrolyte requirements are listed in Table 14-2. Any calculation of maintenance fluid requirements should use the formulas only to estimate a baseline. The actual volume of fluid administered to the patient must be tailored to the patient’s clinical condition and fluid balance. For additional information about fluid and electrolyte balance, see Chapter 12.
Table 14-2 Formulas for Estimating Daily Maintenance Fluid and Electrolyte Requirements for Children
| Daily Requirements | Hourly Requirements | |
| Fluid Requirements Estimated from Weight* | ||
| Newborn (up to 72 h after birth) | 60-100 mL/kg (newborns are born with excess body water) | – |
| Up to 10 kg | 100 mL/kg (can increase up to 150 mL/kg to provide caloric requirements if renal and cardiac function are adequate) | 4 mL/kg |
| 11-20 kg | 1000 mL for the first 10 kg + 50 mL/kg for each kg over 10 kg | 40 mL for first 10 kg + 2 mL/kg for each kg over 10 kg |
| 21-30 kg | 1500 mL for the first 20 kg + 25 mL/kg for each kg over 20 kg | 60 mL for first 20 kg + 1 mL/kg for each kg over 20 kg |
| Fluid Requirements Estimated from Body Surface Area (BSA) | ||
| Maintenance | 1500 mL/m2 BSA | – |
| Insensible losses | 300-400 mL/m2 BSA | – |
| Electrolytes | ||
| Sodium (Na) | 2-4 mEq/kg | – |
| Potassium (K) | 1-2 mEq/kg | – |
| Chloride (Cl) | 2-3 mEq/kg | – |
| Calcium (Ca) | 0.5-3 mEq/kg | – |
| Phosphorous (Phos) | 0.5-2 mmol/kg | – |
| Magnesium (Mg) | 0.4-0.9 mEq/kg | – |
* The “maintenance” fluids calculated by these formulas must only be used as a starting point to determine the fluid requirements of an individual patient. If intravascular volume is adequate, children with cardiac, pulmonary, or renal failure or increased intracranial pressure should generally receive less than these calculated “maintenance” fluids. The formula utilizing body weight generally results in a generous “maintenance” fluid total.
Macronutrient Metabolism and Requirements
Recommended daily requirements of carbohydrate, fat, and protein are summarized in Table 14-3. The percentage of fat required in younger children is usually greater than requirements in older children. Disease states including renal and liver failure may decrease the amount of protein needed.
Table 14-3 Caloric Distribution of Daily Macronutrient Intake
| Macronutrient | Percent of Total Calories (%) |
| Carbohydrates | 40-70 |
| Fat | 20-50 (40-50 in infants) |
| Protein | 7-15 |
Enteral Nutrition
Enteral feeding is preferable to parenteral nutrition, because enteral feedings will maintain gut structure and function and will reduce complications and cost. Specific benefits include intestinal trophism and preservation of the gut barrier to minimize bacterial translocation.38
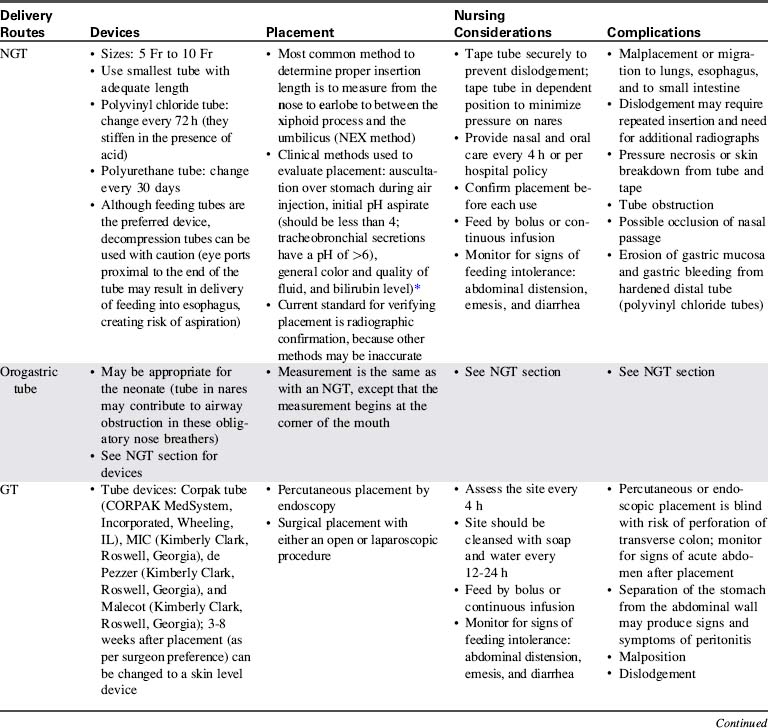
The practice of aspirating residual liquid and refeeding it is controversial. Although a large volume of residual feeding that remains in the stomach may contribute to inadvertent aspiration,45 the residual volume that should be considered significant has not been clearly established. Nurses should measure and record the volume of residual feeding and notify the on-call provider according to unit policy or orders. Enteral feeding delivery devices, routes, placement methods, nursing considerations, and complications are summarized in Table 14-4.

Parenteral Nutrition (PN)
Nursing Responsibilities
The nurse must closely monitor the child’s clinical appearance and fluid and electrolyte balance to prevent and detect complications of PN as soon as possible. This monitoring requires documentation of the quantity and content of the child’s fluid intake and output and evaluation of the child’s fluid and electrolyte status and daily weight. A sample monitoring schedule for children receiving PN is provided in Table 14-5.
Table 14-5 Sample Monitoring Schedule for Children Receiving Parenteral Nutrition: Must Be Tailored to Child’s Clinical Status
| Monitoring | Frequency |
| General and Anthropometric Measurements | |
| Vital signs | Every 4 hours, or more often as patient condition warrants |
| Weight | Daily |
| Strict intake and output | Constant |
| Caloric intake | Daily |
| Height | Weekly |
| Head circumference | Weekly (for children younger than 2 years) |
| Blood Sampling | |
| Glucose | Initial + Daily until stable (more often in neonates) |
| Electrolytes | Initial + Daily until stable |
| BUN, Creatinine | Initial + Weekly |
Ca2+/ionized calcium, 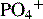 , Mg2+ , Mg2+ | Initial + Weekly (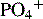 , Mg2+should be checked daily if values not within reference range) , Mg2+should be checked daily if values not within reference range) |
| Alkaline phosphatase, AST, ALT | Initial + Weekly |
| Total and direct bilirubin | Initial + Weekly |
| Total protein, albumin | Initial + Weekly |
| Prealbumin | Initiation |
| Triglycerides, cholesterol | Weekly |
| Zinc, copper, selenium, manganese, iron | Monthly |
| Glucose point-of-care testing | When PN is abruptly discontinued, cycled, or if signs or symptoms of hypoglycemia or hyperglycemia suspected |
| CBC count with differential | When febrile |
| Blood culture | When febrile |
ALT, Alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; Ca2+, calcium; CBC, complete blood count; Mg2+, magnesium; PN, parenteral nutrition; 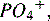 phosphorous.
phosphorous.
Potential complications of PN include electrolyte abnormalities, infection, cholestasis leading to hepatic dysfunction, and hyperlipidemias (with triglyceride administration). In critically ill children, there is an association between hyperglycemia and organ dysfunction.33
Because PN solution contains a high concentration of glucose, it provides an excellent medium for bacterial growth. When hanging a new bag of PN solution and tubing, the nurse must use strict aseptic technique to prevent line contamination. Many hospitals require that the entire tubing system (between PN solution and the patient, including infusion pump tubing) be changed every 24 hours to decrease the possibility of significant bacterial growth. The dressing over the catheter insertion site should be changed when it is no longer occlusive (per hospital policy), using sterile technique and an occlusive dressing. The nurse should assess the catheter insertion site for evidence of inflammation (erythema or exudate) and should report any abnormalities to the appropriate provider. Wound exudate should be cultured and sent for gram stain analysis as ordered (or per protocol). Nurses should be careful to follow the healthcare institutional policies and protocols to prevent central venous catheter-associated blood stream infections (see Chapter 22, Box 22-6 for further information).
Because serum magnesium, phosphate, and calcium levels may fall during PN therapy, providers will monitor the concentration of these elements at least weekly. Trace element deficiencies are more likely to develop with long-term PN therapy or when PN therapy is used in premature infants. The signs and symptoms of copper deficiency include anemia, neutropenia, loss of taste (obviously difficult to assess), and rash. Zinc deficiency can produce an erythematous maculopapular rash (called acrodermatitis enteropathica) over the face, trunk, and digits; poor wound healing; hair loss and loss of taste; and a functional ileus.62 A chromium deficiency can produce a diabetes-like syndrome.62
Because the central venous PN catheter is inserted into a relatively large vein, a loose or cracked tubing connection can rapidly result in significant loss of blood (i.e., hemorrhage). It is important that the nurse check all tubing and catheter connections at least every hour. Because insertion of a central venous catheter creates risk of significant complications, the nurse must ensure that the catheter is secured in place with no possibility of dislodgement. If catheter misplacement or migration is suspected, a chest radiograph may be needed to verify placement (for further information, see Chapter 10).
Cholestatic jaundice is a serious but incompletely understood complication of PN that is associated with periportal fibrosis, bile duct proliferation, and bile stasis. This complication contributes to morbidity and mortality in younger infants and children with short gut syndrome (SGS) following intestinal resection.10 The first sign of cholestatic jaundice is elevation in concentrations of liver enzymes (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]). In addition, the bilirubin will begin to rise and the child may appear jaundiced; cholestasis is defined as a direct bilirubin greater than 2 mg/dL. Risk of cholestatic jaundice can be reduced by cycling and limiting lipid administration to 1 g/kg per day or using alternate day dosing of the fat emulsion. If cholestasis is present, treatment includes administration of ursodeoxycholic acid.
Common clinical conditions
Intestinal Failure
Pathophysiology
Each child with intestinal failure is unique, because there are no absolute criteria for the amount and type of bowel needed to sustain absorption of nutrients for appropriate growth. The normal estimated bowel length at birth is 250 ± 40 cm.22 After the loss of a bowel segment, the intestine undergoes a process termed intestinal adaptation. As a general rule, infants can experience acceptable intestinal function with less than 15 cm of intestine if the ileocecal valve is intact, and with 30 to 45 cm of intestine if the ileocecal valve is absent or does not function.17 It is unlikely that children with less than 20 cm of remaining bowel will be able to grow and develop with enteral nutrition alone. Intestinal transplantation may offer hope to these patients.
Intestinal adaptation is characterized by increasing intestinal mass, lengthening of villi, and improved absorption at the epithelial level.10,22 This process allows for the remaining intestine to compensate for the loss of the bowel by increasing its surface area and functional abilities.10 Successful adaptation is described as the ability to achieve normal growth, fluid balance, and electrolyte concentration without PN.53 The time frame required for adaptation is unclear and dependent on the etiology of the SBS and the functional state of the remaining bowel, although adaptation can occur over weeks to months.10,22 Children have been transitioned to enteral feedings exclusively over periods as long as 8 years. There are a number of metabolic derangements that occur after loss of bowel that are summarized in Table 14-6.
Table 14-6 Metabolic Derangements and Consequences in Children with Short Gut Syndrome
| Derangements | Consequences |
| Early | |
| Gastric hypersecretion | Peptic ulceration |
| Dumping syndrome | Diarrhea, hyperglycemia, reactive hypoglycemia |
| Rapid intestinal transit | Nutrient malabsorption |
| High output from enterostomies | Electrolyte disturbances |
| Late | |
| Bile and fatty acid malabsorption | Gallstones, steatorrhea |
| Bowel dilation and stasis | Bacterial overgrowth syndrome, D-lactic acidosis |
| Anastomotic ulceration | Gastrointestinal bleeding |
From Cohran VC and Kocoshis SA: Short bowel. In Baker S, Baker R, David A, editors: Pediatric nutrition support, Sudburry, MA, 2007, Jones and Bartlett.
Management
Often 20 Kcal/ounce feedings are initiated and can be advanced to higher caloric density (to as high as 30 Kcal/ounce [1 Kcal/mL]) as tolerated. Usually these children are fed with casein hydrolysate formulas and elementary amino acid-based formulas such as Pregestimil (Mead Johnson, Evansville, IN), EleCare (Abbott Nutrition, Columbus, OH), and Neocate (Nutricia North America, Gaithersburg, MD) in infants and Peptamen Jr (Nestle Nutrition, North America, Minneapolis, MN) and Neocate Jr.(Nutricia North America, Gaithersburg, MD) in older children. These formula types are easier to digest and are less likely to trigger an immune response, so they will enhance bowel adaptation.10
When children are unable to achieve appropriate growth with enteral feedings, they may be considered surgical candidates for procedures to restore normal bowel diameter, lengthen the bowel, or both. The goals of surgical intervention include reducing stasis, improving motility, and increasing the effective mucosal surface area.29 The most common short bowel procedures are the Bianchi, Kimura, and the serial transverse enteroplasty procedure.
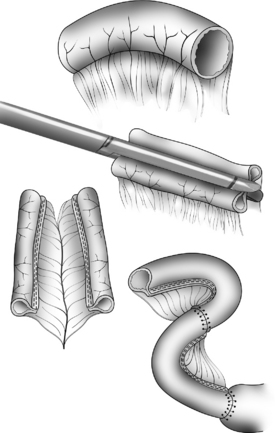
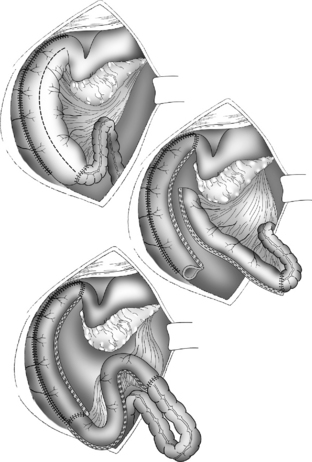
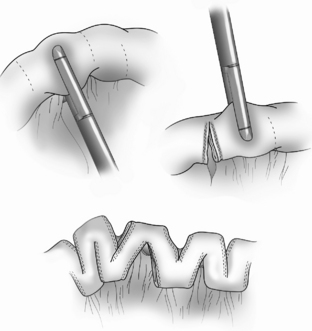
The Bianchi procedure is the oldest procedure and involves a longitudinal incision to create two tubes to lengthen the bowel (Fig. 14-5). The Kimura procedure (Fig. 14-6) is an alternative procedure for patients with SGS and inadequate mesentery who are not candidates for the Bianchi procedure. The serial transverse enteroplasty (STEP) procedure augments bowel length by stapling dilated bowel in a zigzag fashion to achieve more effective bowel surface area (Fig. 14-7).
With advances in surgical techniques and immunosuppressive therapy, intestine transplantation is successful as an isolated procedure and when combined with transplantation of other organs.54 A major reason for the increased success of intestinal transplantation is the availability of more potent immunosuppressants such as tacrolimus (Prograf). The major limiting factor that prevents widespread use of transplantation is the lack of available organs for transplantation. Because children with less than 15 cm of bowel are not likely to tolerate enteral feeding and thrive, they should be referred to an intestinal rehabilitation and transplant program as soon as possible.
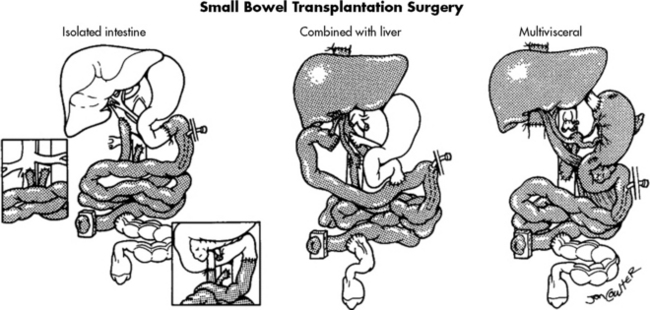
The ideal intestine donor has the same blood type, weighs within 10% of the recipient’s body weight, and is close in age to the recipient. All intestine recipients have a stoma to allow for bowel surveillance and access for endoscopy and biopsy. Figure 14-8 shows the technical details of isolated intestine transplant procedure and intestine transplant in combination with other organs.54
Critical care goals are to support the patient to be hemodynamically stable, with adequate oxygenation and ventilation, free of requirement for mechanical ventilation support. Airway clearance and spontaneous ventilation may be ineffective as a result of the lengthy abdominal surgical procedure (and resultant need for anesthetic administration during the long procedure), visceral edema, and pain. With the postoperative resolution of coagulopathies, aggressive chest physiotherapy is initiated. Intestine recipients generally require mechanical ventilation longer than isolated liver recipients (see Liver Transplantation in Chapter 17), because intestine recipients typically are in poor health before the transplant and often have a precarious fluid balance. Additional challenges include open abdominal wounds and potential need for surgical reexploration for complications such as perforation or bleeding.
Post-transplant lymphoproliferative disorder (PTLD) is one of the most underrated complications of immunosuppression. PTLD is the development of continually proliferating B-lymphocytes, presumably under the influence of Epstein-Barr virus. Diagnosis is made by clinical examination and histologic review. Clinical signs and symptoms include fever, lymphadenopathy, GI symptoms, and weight loss. If the biopsy specimen is diagnostic for PTLD, treatment includes holding or reducing immunosuppression and initiating IV antiviral therapy and immunoglobulin therapy (see Chapter 17 for additional information).
Gastrointestinal Bleeding
Etiology and Pathophysiology
GI bleeding in children can result from inflammation of the intestine, congenital or acquired visceral or vascular anomalies, trauma, esophageal varices, ulcers, or coagulopathies. The incidence of acquired GI bleeding in critically ill children receiving mechanical ventilation has been reported to be as high as 51.8% in some series.14,47 Identified risk factors include a pediatric risk of mortality 2 score of 10 or higher, operating room procedures longer than 3 hours, hepatic insufficiency, coagulopathy, respiratory failure, and high-pressure ventilator settings of greater than or equal to 25 cm H2O.14,47
Lower GI bleeding is the result of mucosal disruption distal to the ligament of Treitz and can be caused by Crohn’s disease, intussusception, or ischemic injury. Common causes of GI bleeding in children are listed in Table 14-7.
Table 14-7 Causes of Gastrointestinal Bleeding in Infants and Children
| Age Group and Status | Upper GI Bleeding | Lower GI Bleeding |
| Healthy neonate | Swallowed maternal blood, hemorrhagic disease of the newborn, esophagitis, gastric duplication | Swallowed maternal blood, infectious colitis, milk allergy, hemorrhagic disease of the newborn, duplication of the bowel, Meckel’s diverticulum, anal fissure |
| Sick neonate | Stress ulcer, gastritis, vascular malformations | Necrotizing enterocolitis, infectious colitis, disseminated coagulopathy, midgut volvulus, intussusception |
| Infancy | Stress ulcer, esophagitis, gastritis, gastric duplication | Anal fissure, infectious colitis, milk allergy, nonspecific colitis, juvenile polyps, intestinal duplication, intussusception, Meckel’s diverticulum |
| Preschool age | Esophagitis, gastritis, stress ulcer, peptic ulcer disease, foreign body, caustic ingestion, vascular disease (Rendu-Osler-Weber disease, hemophilia), trauma, portal hypertension | Infectious colitis, juvenile polyps, anal fissure, intussusception, Meckel’s diverticulum, angio-dysplasia, Henoch-Schönlein purpura, hemolytic-uremic syndrome, inflammatory bowel disease |
| School age and adolescence | Esophagitis, gastritis, stress ulcer, peptic ulcer disease, portal hypertension, trauma | Infectious colitis, inflammatory bowel disease, polyps, angiodysplasia, Henoch-Schönlein purpura, hemolytic-uremic syndrome, hemorrhoids, rectal trauma |
GI, Gastrointestinal.
Data from Arensman RM, Browne M, Madonna MB: Gastrointestinal bleeding. In Grosfeld JL et al, editors: Pediatric surgery, ed 6, Philadelphia, 2006, Mosby; Martin SA, Simone S: Gastrointestinal system: In Slota MC, editor: Core curriculum for pediatric critical care nursing, ed 2, St Louis, 2006, Mosby-Elsevier.
Microscopic bleeding may cause no symptoms and may be detectable only through analysis of GI secretions or feces. Significant GI bleeding may result in hypovolemia and low cardiac output, shock, and death (see also Shock, in Chapter 6). See the section on Gastrointestinal Bleeding in the Chapter 14 Supplement on the Evolve Website for additional information about the pathophysiology of GI hemorrhage.
Clinical Signs and Symptoms
Metabolic acidosis and a rise in serum lactate may be noted. Oxyhemoglobin desaturation may not be present or detected by pulse oximetry, because existing hemoglobin may be saturated with oxygen. The oximeter device may have difficulty detecting a signal if the child’s pulses are weak. The nurse should notify an on-call provider immediately of these findings, because the patient’s status is critical (see Chapter 6 for more information about recognition and treatment of shock).
During the first days of life, the Apt test (named for Leonard Apt) may be performed to distinguish between swallowed maternal blood and GI bleeding as a cause of blood in the newborn’s stool.4 The Apt test is performed by placing blood from the neonate on filter paper with 1% sodium hydroxide (a reagent that reacts with fetal hemoglobin).4 Maternal blood will appear rusty brown, whereas the neonate’s blood that contains fetal hemoglobin will remain pink or red.4 The pink or red color is a positive result, indicative of presence of blood from the neonate.
Management
The three phases of management of the child with GI bleeding are resuscitation, specific diagnosis, and specific treatment. A diagnostic algorithm for upper GI bleeding is presented in Fig. 14-9.
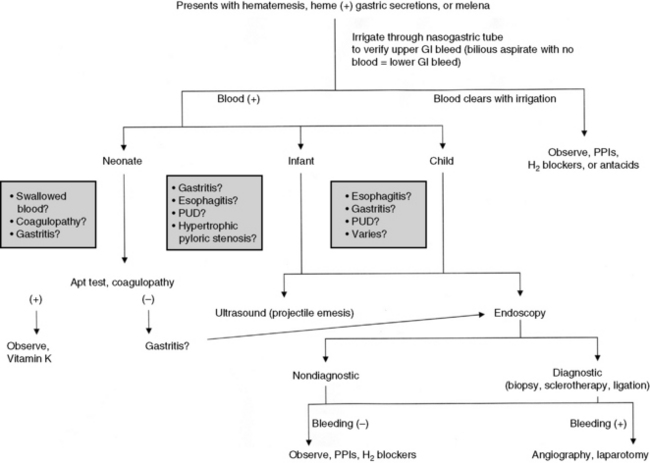
During resuscitation and replacement of intravascular volume, nursing observations may help to determine the source of the child’s bleeding. If saline lavage through a nasogastric tube reveals grossly bloody or red-tinged aspirate, ongoing upper intestinal bleeding is present. Nursing interventions during resuscitation of the child with GI bleeding are summarized in Box 14-1.
Box 14-1 Nursing Interventions During Resuscitation of the Child with GI Bleeding
1. Assess and support airway, breathing, and oxygenation. Optimize oxygenation and provide supplementary oxygen as indicated. If the child’s level of consciousness is decreased, the child will likely require insertion of an advanced airway and support of ventilation.
2. Restore adequate intravascular volume. Establish vascular (intravascular or intraosseous) access. Because the rate of intravenous fluid replacement will be limited by the size of the vascular catheter, insert the largest catheter possible. It is preferable to establish two vascular catheters to allow one catheter to be used for rapid volume expansion while the other is used for administering medication and measuring the child’s CVP.
3. Assess systemic perfusion. Monitor arterial and central venous pressures and systemic perfusion.
4. Insert a urinary catheter to monitor urine output.
5. Obtain blood samples for frequent (at least every 4 h) measurement of hematocrit or complete blood count; notify an on-call provider if either falls.
6. Administer blood volume expanders or packed red blood cells as ordered. Children with active gastrointestinal bleeding should have an active type and cross match with blood available at all times.
7. Assess for signs of further hemorrhage, including signs of poor systemic perfusion, abdominal pain or tenderness, changes in bowel sounds, hematemesis, or hematochezia.
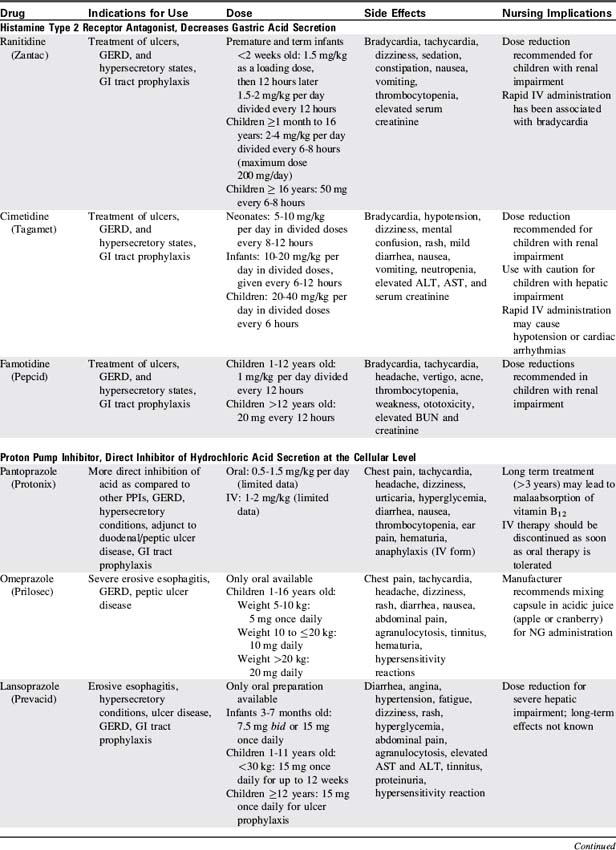
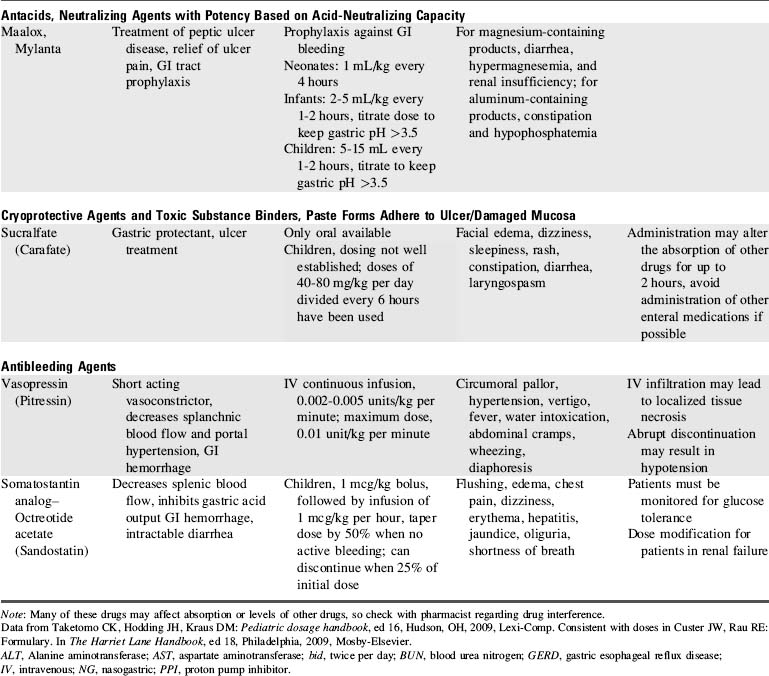
8. Administer antibleeding pharmacologic agents as prescribed. See Table 14-8 for specific drug information.
Proton pump inhibitors and histamine-2 receptor antagonists are the most commonly used pharmacologic agents to prevent the development of GI bleeding (Table 14-8). Although only a small number of critically ill children will develop gastric bleeding, the associated morbidity justifies the common practice of prophylaxis.
Hyperbilirubinemia
Etiology
When the neonate’s TSB is elevated, the bilirubin can cross the blood-brain barrier, causing acute bilirubin encephalopathy or the chronic form of bilirubin encephalopathy, kernicterus. Kernicterus is a yellow staining and brain tissue damage with degenerative lesions, resulting from central nervous system exposure to high concentrations of unconjugated bilirubin. Hyperbilirubinemia is commonly associated with prematurity, breast feeding, and other factors summarized in Box 14-2.
Box 14-2 Risk Factors for Hyperbilirubinemia in the Neonate
Factors That Increase Risk
• Jaundice in first 24 h (usually related to hemolysis)
• Visible jaundice before hospital discharge or TSB or TcB level in the high-risk zone
• Hemolytic disease: blood group incompatibility, Rh incompatibility, glucose-6-phosphate dehydrogenase deficiency
• Prematurity (less than 35 weeks’ gestation)
• Cephalohematoma or significant bruising
• Sibling previously received phototherapy (possible genetic cause)
• Ethnicity: East Asian, certain Native American tribes, and Greek ancestry
• Maternal age greater than 25 years
• Macrosomic infants of diabetic mothers
Data from American Academy of Pediatrics Subcommittee on Hyperbilirubinemia: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297, 2004; Watson RL: Hyperbilirubinemia. Crit Care Nurs Clin N Am 21:97, 2009.Rh, Rhesus (factor); TcB, transcutaneous bilirubin; TSB, total serum bilirubin.
In the first week of life, it is estimated that approximately 60% of normal newborns will become clinically jaundiced.42 Newborns are now discharged from the hospital at approximately 24 to 48 hours after birth, before the typical TSB peak at 48 to 96 hours. As a result, hyperbilirubinemia is the most common reason for hospital readmission of the neonate. A sentinel event alert was issued by the Joint Commission in 2001, because an increased number of cases of kernicterus were being diagnosed in otherwise healthy newborns.
Pathophysiology
When RBCs reach the end of their 120-day life span, they normally are sequestered in the spleen. The cells are destroyed, and the heme portion of the hemoglobin molecule is oxidized, and bilirubin is formed. Bilirubin is bound to albumin in the plasma and taken up in the liver, where it is combined with a sugar through the action of the enzyme, bilirubin uridine diphosphate glucuronosyltransferase, making conjugated bilirubin.11 Conjugated bilirubin is water soluble, it cannot cross the blood-brain barrier, and it is normally excreted in bile (Fig. 14-10). Free bilirubin, called unconjugated (indirect) bilirubin, is lipid soluble and not water soluble. Because unconjugated bilirubin is thought to diffuse freely into the brain, high concentrations of this form of bilirubin may be neurotoxic and cause kernicterus.
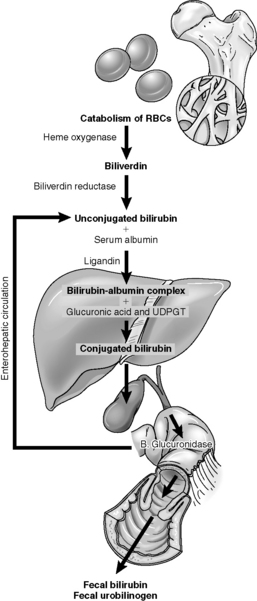
Fig. 14-10 The pathophysiology of neonatal hyperbilirubinemia.
(From Colletti JE, et al: An emergency medicine approach to neonatal hyperbilirubinemia. Emerg Med Clin N Am 25:1117-1135, 2007.)
Kernicterus is yellow (bilirubin) staining of the basal ganglia in the brain of neonates with severe jaundice. Although the precise mechanisms responsible for the entry of bilirubin into the brain are not known, disruption of the blood brain barrier from increased permeability (e.g., caused by hyperosmolarity or severe asphyxia), prolonged transit time (e.g., caused by increased central venous pressure [CVP]), or increased blood flow (e.g., caused by hypercarbia and acidosis) are thought to be contributory.63
The neurotoxic sequelae of hyperbilirubinemia are the most worrisome, and a grading for acute bilirubin encephalopathy has been proposed. The earliest signs may include alteration in the tone of extensor muscles (hypotonia or hypertonia), retrocollis (backward arching of the neck), opisthotonus (backward arching of the trunk), and a poor suck.3 The early symptoms may intensify and be accompanied by a shrill cry and unexplained irritability alternating with lethargy.3 Therapy at this stage may prevent advancement of symptoms. Cessation of feeding, irritability, seizures, and altered mental status are later symptoms of acute bilirubin encephalopathy. The final stage is kernicterus and may be irreversible with the development of cerebral palsy, deafness or hearing loss, and impairment of upward gaze. Kernicterus has a significant mortality (at least 10%) and long-term morbidity (at least 70%), and most cases are reported in infants with TSB greater than 20 mg/dL.28
Noninvasive (transcutaneous) bilirubin measurements can be obtained with newer instruments. Published data suggest that in most infants the measurements are within 2 to 3 mg/dL of serum measurements and can replace TSB measurements in most cases.2 In infants undergoing phototherapy, this measurement is not reliable and should not be used.
It is possible to measure serum levels of total bilirubin and direct (or conjugated) bilirubin. Current recommendations caution against inferring the indirect (unconjugated) bilirubin from the TSB, because the difference between the total and direct bilirubin is not constant. Current guidelines recommend that providers evaluate the TSB level63; the higher the TSB, the greater the risk of bilirubin encephalopathy. Treatment is indicated for infants with bilirubin levels in excess of 20 mg/dL.
Management
The current American Academy of Pediatrics (AAP) recommendations for phototherapy are based on age and TSB levels (see Evolve Fig. 14-1 in the Chapter 14 Supplement on the Evolve Website to view a graphic representation of recommended therapies based on infant age and total serum bilirubin concentration). Prophylactic phototherapy is often instituted for neonates weighing less than 1500 g. Although exclusive breast feeding can contribute to high TSB levels, the AAP recommends that breast feeding be continued with 10 to 12 feedings recommended per day, plus administration of supplementary intravenous fluid to treat dehydration.2
Phototherapy converts bilirubin to a water-soluble form that can be excreted without glucuronidation. Phototherapy units contain day lights, fluorescent tubes, fiberoptic light, and halogen bulbs. The effectiveness of phototherapy is determined by the type of light, the infant’s distance to the light, and the amount of exposed body surface area. Light emitted at a wavelength in the blue-green spectrum of 425 to 495 nm is thought to be most effective because of the optical qualities of bilirubin and the skin. The AAP recommends the use of special blue fluorescent lamps or light-emitting diodes.41,42
The duration of phototherapy required is affected by the TSB level, the cause of the hyperbilirubinemia, and the infant’s age. For infants readmitted for phototherapy during the first days of life, the treatment is usually discontinued when TSB levels reach 13 to 14 mg/dL. It is not uncommon for infants to experience a rebound increase in TSB level of 1 to 2 mg/dL when the therapy is discontinued.41
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


