CHAPTER 21 In people with PD, neurotransmission is disrupted primarily in the striatum, an important component of the extrapyramidal system. A simplified model of striatal neurotransmission is depicted in Figure 21–1A. As indicated, proper function of the striatum requires a balance between two neurotransmitters: dopamine and acetylcholine (ACh). Dopamine is an inhibitory transmitter; ACh is excitatory. According to the model, the neurons that release dopamine inhibit neurons that release gamma-aminobutyric acid (GABA, another inhibitory transmitter). In contrast, the neurons that release ACh excite the neurons that release GABA. Movement is normal when the inhibitory influence of dopamine and the excitatory influence of ACh are in balance. Note that the neurons that supply dopamine to the striatum originate in the substantia nigra. Between 70% and 80% of these neurons must be lost before PD becomes clinically recognizable. This loss takes place over 5 to 20 years. Put another way, neuronal degeneration begins long before overt motor symptoms appear. In PD, there is an imbalance between dopamine and ACh in the striatum (Fig. 21–1B). As noted, the imbalance results from degeneration of the neurons that supply dopamine to the striatum. In the absence of dopamine, the excitatory influence of ACh goes unopposed, causing excessive stimulation of the neurons that release GABA. Overactivity of these GABAergic neurons contributes to the motor symptoms that characterize PD. As discussed in Chapter 31, movement disorders similar to those of PD can occur as side effects of antipsychotic drugs. These dyskinesias, which are referred to as extrapyramidal side effects, result from blockade of dopamine receptors in the striatum. This drug-induced parkinsonism can be managed with some of the drugs used to treat PD. As shown in Table 21–1, dopaminergic drugs act by several mechanisms: levodopa promotes dopamine synthesis; the dopamine agonists activate dopamine receptors directly; inhibitors of monoamine oxidase-B (MAO-B) prevent dopamine breakdown; amantadine promotes dopamine release (and may also block dopamine reuptake); and the inhibitors of catechol-O-methyltransferase (COMT) enhance the effects of levodopa by blocking its degradation. TABLE 21–1 Dopaminergic Agents for Parkinson’s Disease COMT = catechol-O-methyltransferase, DA = dopamine, MAO-B = type B monoamine oxidase. • Initiation and Treatment for Parkinson Disease (Updated), 2002 • Diagnosis and Prognosis for New Onset Parkinson Disease, 2006 • Neuroprotective Strategies and Alternative Therapies for New Onset Parkinson Disease, 2006 • Evaluation and Treatment of Depression, Psychosis and Dementia in Parkinson Disease, 2006 • Medical and Surgical Treatment of Parkinson Disease with Motor Fluctuations and Dyskinesia, 2006 The entire set is available free online at www.neurology.org. The recommendations below are based on these guidelines. Abrupt loss of effect, often referred to as the “on-off” phenomenon, can occur at any time during the dosing interval—even while drug levels are high. “Off” times may last from minutes to hours. Over the course of treatment, “off” periods are likely to increase in both intensity and frequency. Drugs that can help reduce “off” times are listed in Table 21–2. As discussed below, avoiding high-protein meals may also help. TABLE 21–2 Drugs for Motor Complications of Levodopa Therapy COMT = catechol-O-methyltransferase, DA = dopamine, MAO-B = type B monoamine oxidase. Levodopa reduces symptoms by increasing synthesis of dopamine in the striatum (Fig. 21–2). Levodopa enters the brain via an active transport system that carries it across the blood-brain barrier. Once in the brain, the drug undergoes uptake into the few dopaminergic nerve terminals that remain in the striatum. Following uptake, levodopa, which has no direct effects of its own, is converted to dopamine, its active form. As dopamine, levodopa helps restore a proper balance between dopamine and ACh. Conversion of levodopa to dopamine is depicted in Figure 21–3. As indicated, the enzyme that catalyzes the reaction is called a decarboxylase (because it removes a carboxyl group from levodopa). The activity of decarboxylases is enhanced by pyridoxine (vitamin B6). Why is PD treated with levodopa and not with dopamine itself? There are two reasons. First, dopamine cannot cross the blood-brain barrier (see Fig. 21–2). As noted, levodopa crosses the barrier by means of an active transport system, a system that does not transport dopamine itself. Second, dopamine has such a short half-life in the blood that it would be impractical to use even if it could cross the blood-brain barrier. Ironically, levodopa, which is given to alleviate movement disorders, actually causes movement disorders in many patients. About 80% develop involuntary movements within the first year. Some dyskinesias are just annoying (eg, head bobbing, tics, grimacing), whereas others can be disabling (eg, ballismus, choreoathetosis). These dyskinesias develop just before or soon after optimal levodopa dosage has been achieved. Dyskinesias can be managed in three ways. First, the dosage of levodopa can be reduced. However, dosage reduction may allow PD symptoms to re-emerge. Second, we can give amantadine (see below), which can reduce dyskinesias in some patients. If these measures fail, the only remaining options are surgery and electrical stimulation (see Box 21–1). SURGICAL AND ELECTRICAL TREATMENTS FOR PARKINSON’S DISEASE Posteroventral medial pallidotomy, or simply pallidotomy, is a neurosurgical procedure for destroying a region of the globus pallidus. As indicated in Figure 21–1, the globus pallidus, which helps regulate movement, receives input from the striatum. In patients with PD, striatal input to the globus pallidus is disrupted, causing the globus pallidus itself to malfunction. It has been argued that altered output from the globus pallidus underlies many of the symptoms of PD, including tremor, rigidity, and bradykinesia. The results of pallidotomy support this argument: For many patients with PD, unilateral destruction of the posteroventral medial region of the globus pallidus produces a substantial improvement in symptoms. The most consistent benefit is a reduction in levodopa-induced dyskinesias. Motor control during levodopa “off” times also improves. In contrast, very little improvement is seen during levodopa “on” times. Following pallidotomy, about 50% of patients who previously needed help with activities of daily living are able to live independently. Pallidotomy may also permit a temporary reduction in levodopa dosage. Although complications of the procedure are generally mild, intracerebral hemorrhage is a potential and serious risk. Two second-generation antipsychotics—clozapine and quetiapine—have been used successfully to manage levodopa-induced psychosis. Unlike the first-generation antipsychotic drugs, clozapine and quetiapine cause little or no blockade of dopamine receptors in the striatum, and hence do not cause EPS. In patients taking levodopa, these drugs can reduce psychotic symptoms without intensifying symptoms of PD. Interestingly, the dosage of clozapine is only 25 mg/day, about 20 times lower than the dosage used for schizophrenia. Clozapine and quetiapine are discussed at length in Chapter 31. Interactions between levodopa and other drugs can (1) increase beneficial effects of levodopa, (2) decrease beneficial effects of levodopa, and (3) increase toxicity from levodopa. Major interactions are summarized in Table 21–3. Several interactions are discussed immediately below; others are discussed later on. TABLE 21–3 Major Drug Interactions of Levodopa *First-generation antipsychotic agents block dopamine receptors in the striatum and can thereby nullify the therapeutic effects of levodopa. Two second-generation antipsychotics—clozapine [Clozaril] and quetiapine [Seroquel]—do not block dopamine receptors in the striatum, and hence do not nullify the therapeutic effects of levodopa.
Drugs for parkinson’s disease
Pathophysiology that underlies motor symptoms
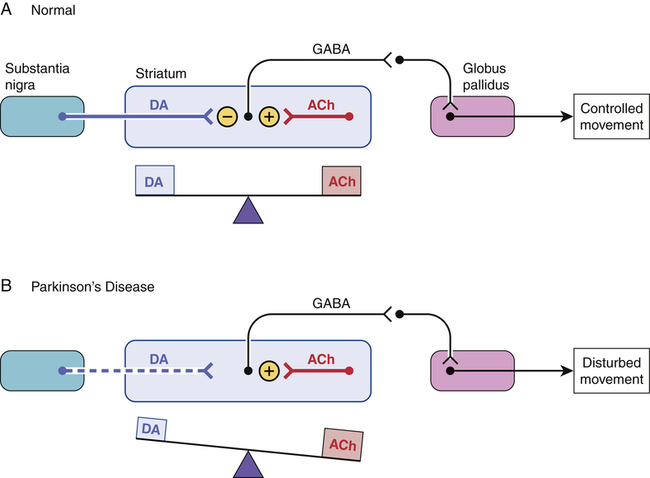
 A model of neurotransmission in the healthy striatum and parkinsonian striatum.
A model of neurotransmission in the healthy striatum and parkinsonian striatum.
A, In the healthy striatum, dopamine (DA) released from neurons originating in the substantia nigra inhibits the firing of neurons in the striatum that release gamma-aminobutyric acid (GABA). Conversely, neurons located within the striatum, which release acetylcholine (ACh), excite the GABAergic neurons. Hence, under normal conditions, the inhibitory actions of DA are balanced by the excitatory actions of ACh, and controlled movement results. B, In Parkinson’s disease, the neurons that supply DA to the striatum degenerate. In the absence of sufficient DA, the excitatory effects of ACh go unopposed, and disturbed movement results.
Overview of motor symptom management
Drugs employed

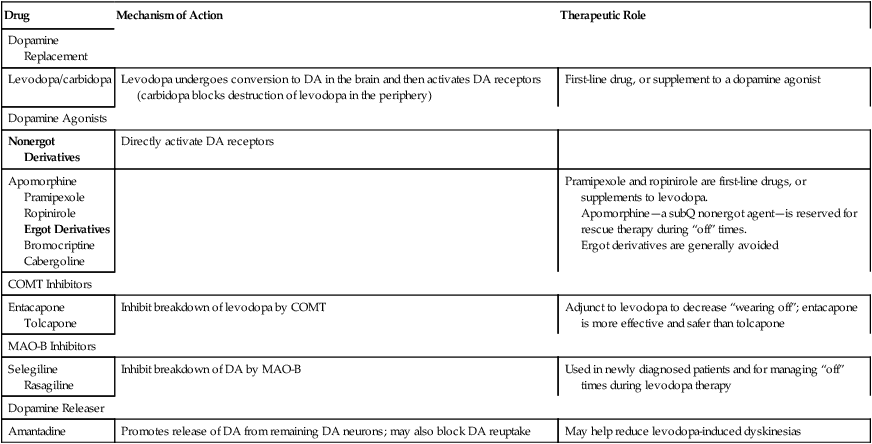
Drug
Mechanism of Action
Therapeutic Role
Dopamine Replacement
Levodopa/carbidopa
Levodopa undergoes conversion to DA in the brain and then activates DA receptors (carbidopa blocks destruction of levodopa in the periphery)
First-line drug, or supplement to a dopamine agonist
Dopamine Agonists
Nonergot Derivatives
Directly activate DA receptors
Apomorphine
Pramipexole
Ropinirole
Ergot Derivatives
Bromocriptine
Cabergoline
Pramipexole and ropinirole are first-line drugs, or supplements to levodopa.
Apomorphine—a subQ nonergot agent—is reserved for rescue therapy during “off” times.
Ergot derivatives are generally avoided
COMT Inhibitors
Entacapone
Tolcapone
Inhibit breakdown of levodopa by COMT
Adjunct to levodopa to decrease “wearing off”; entacapone is more effective and safer than tolcapone
MAO-B Inhibitors
Selegiline
Rasagiline
Inhibit breakdown of DA by MAO-B
Used in newly diagnosed patients and for managing “off” times during levodopa therapy
Dopamine Releaser
Amantadine
Promotes release of DA from remaining DA neurons; may also block DA reuptake
May help reduce levodopa-induced dyskinesias
Clinical guidelines
Pharmacology of the drugs used for motor symptoms
Levodopa
Use in parkinson’s disease
Acute loss of effect.

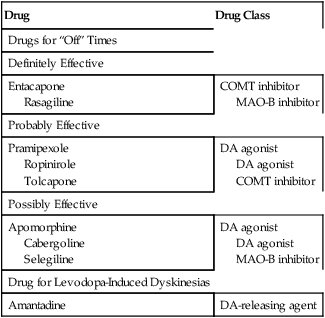
Drug
Drug Class
Drugs for “Off” Times
Definitely Effective
Entacapone
Rasagiline
COMT inhibitor
MAO-B inhibitor
Probably Effective
Pramipexole
Ropinirole
Tolcapone
DA agonist
DA agonist
COMT inhibitor
Possibly Effective
Apomorphine
Cabergoline
Selegiline
DA agonist
DA agonist
MAO-B inhibitor
Drug for Levodopa-Induced Dyskinesias
Amantadine
DA-releasing agent
Mechanism of action
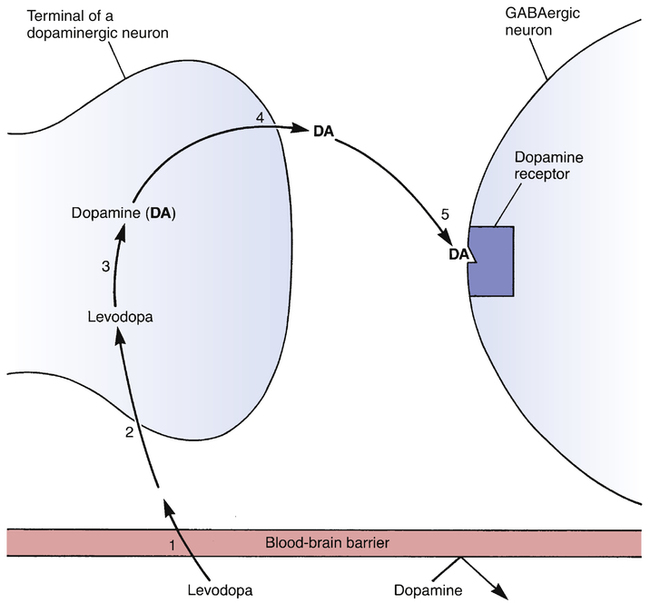
 Steps leading to alteration of CNS function by levodopa.
Steps leading to alteration of CNS function by levodopa.
To produce its beneficial effects in PD, levodopa must be (1) transported across the blood-brain barrier; (2) taken up by dopaminergic nerve terminals in the striatum; (3) converted into dopamine; (4) released into the synaptic space; and (5) bound to dopamine receptors on striatal GABAergic neurons, causing them to fire at a slower rate. Note that dopamine itself is unable to cross the blood-brain barrier, and hence cannot be used to treat PD.
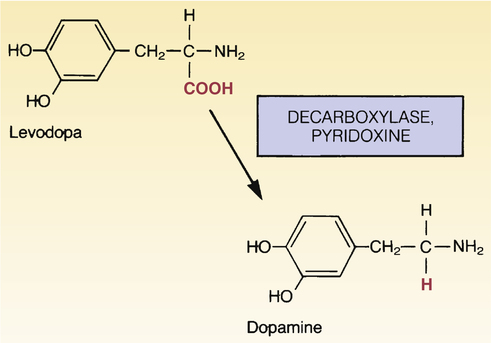
 Conversion of levodopa to dopamine.
Conversion of levodopa to dopamine.
Decarboxylases present in the brain, liver, and intestine convert levodopa into dopamine. Pyridoxine (vitamin B6) accelerates the reaction.
Adverse effects
Dyskinesias.
 BOX 21–1
BOX 21–1  SPECIAL INTEREST TOPIC
SPECIAL INTEREST TOPIC
Psychosis.
Drug interactions

Drug Category
Drug
Mechanism of Interaction
Drugs that increase beneficial effects of levodopa
Carbidopa
Entacapone, tolcapone
Inhibits peripheral decarboxylation of levodopa
Inhibit destruction of levodopa by COMT in the intestine and peripheral tissues
Apomorphine, bromocriptine, cabergoline, pramipexole, ropinirole
Stimulate dopamine receptors directly, and thereby add to the effects of dopamine derived from levodopa
Amantadine
Promotes release of dopamine
Anticholinergic drugs
Block cholinergic receptors in the CNS, and thereby help restore the balance between dopamine and ACh
Drugs that decrease beneficial effects of levodopa
Antipsychotic drugs*
Block dopamine receptors in the striatum
Drugs that increase levodopa toxicity
MAO inhibitors (especially nonselective MAO inhibitors)
Inhibition of MAO increases the risk of severe levodopa-induced hypertension
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Drugs for parkinson’s disease
Only gold members can continue reading. Log In or Register to continue
Get Clinical Tree app for offline access
