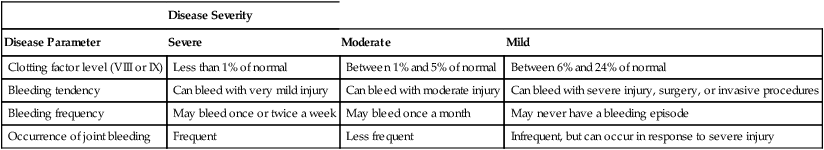
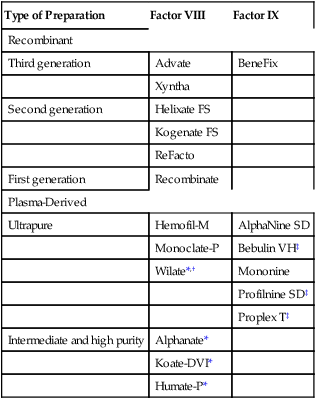
CHAPTER 54 In people with hemophilia, there is a failure of hemostasis, the process by which bleeding is stopped. As discussed in Chapter 52, hemostasis occurs in two stages: (1) formation of a platelet plug followed by (2) production of fibrin, a protein that reinforces the platelet plug. In patients with hemophilia, platelet aggregation proceeds normally, but fibrin production does not. The underlying problem is a deficiency of clotting factors—specifically, factor VIII (in hemophilia A) and factor IX (in hemophilia B). As indicated in Figure 54–1, both factors are part of the contact activation (intrinsic) coagulation pathway, and both—in their activated forms—are needed to catalyze the conversion of factor X to its active form (factor Xa), which in turn catalyzes the conversion of prothrombin to thrombin, which catalyzes the formation of fibrin. If either factor VIII or factor IX is deficient, the contact activation pathway will not work properly, causing clot formation to be delayed. As a result, bleeding will continue longer than in the population at large. Hemophilia may be severe, moderate, or mild, depending on the degree of clotting factor deficiency. Patients with severe hemophilia may experience life-threatening hemorrhage in response to minor trauma, whereas those with mild hemophilia may experience little or no excessive bleeding. The defining characteristics of severe, moderate, and mild hemophilia are summarized in Table 54–1. TABLE 54–1 Clinical Classification of Hemophilia Severity In some patients receiving factor VIII or factor IX, antibodies against the factor develop. These antibodies, referred to as inhibitors, prevent the factor from working. When inhibitors are present, bleeding can be stopped by infusing activated factor VII. Other treatments are also available, as discussed below under Managing Patients Who Develop Inhibitors. What about the second-generation NSAIDs, known as cyclooxygenase-2 (COX-2) inhibitors? As discussed in Chapter 71, the COX-2 inhibitors (eg, celecoxib) do not suppress platelet aggregation, and they cause less GI ulceration and bleeding than traditional NSAIDs. Accordingly, these agents are clearly preferred to traditional NSAIDs, although their safety in hemophilia has not been proved. Children with hemophilia should undergo the normal immunization schedule (see Chapter 68). Some clinicians inject vaccines subQ, rather than IM, to avoid muscle hemorrhage. However, since the efficacy of subQ vaccination is not certain, and since most patients tolerate IM injections without bleeding, IM vaccination is generally preferred. The risk of bleeding after IM injection can be reduced by prolonged application of pressure. Factor VIII concentrates are made in two basic ways: (1) purification from human plasma and (2) production in cell culture using recombinant DNA technology. Recombinant factor VIII is somewhat safer than plasma-derived factor VIII, but is also more expensive. All factor VIII products, whether recombinant or plasma derived, are equally effective. Available products are listed in Table 54–2. TABLE 54–2 Some Factor VIII and Factor IX Concentrates *Contains von Willebrand factor in addition to factor VIII. †Approved only for von Willebrand disease. As indicated in Table 54–2, plasma-derived factor VIII is available in varying degrees of purity. The ultrapure products (eg, Hemofil-M) are prepared using monoclonal antibodies.
Drugs for hemophilia
Basic considerations
Pathophysiology
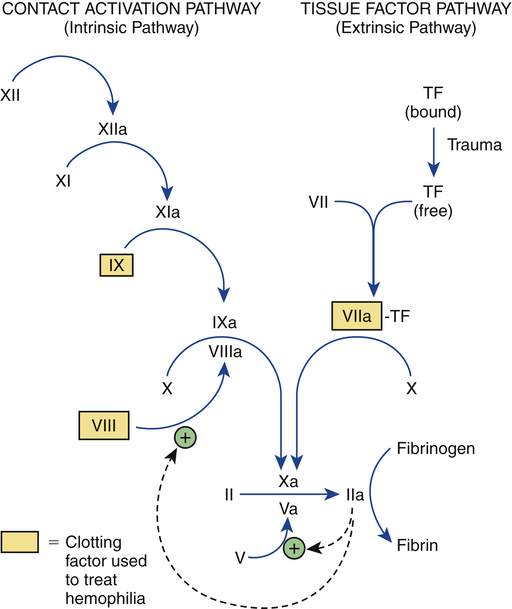
 Outline of the coagulation cascade showing clotting factors used to treat hemophilia.
Outline of the coagulation cascade showing clotting factors used to treat hemophilia.
TF = tissue factor. Common names for factors shown in roman numerals: II = prothrombin, IIa = thrombin, VII = proconvertin, VIII = antihemophilic factor, IX = Christmas factor, X = Stuart factor, XI = plasma thromboplastin antecedent, and XII = Hageman factor. The letter “a” after a factor’s name (eg, factor VIIIa) indicates the active form of the factor. Note that factors VIII and IX, which are deficient in hemophilia A and B, respectively, are part of the contact activation (intrinsic) coagulation pathway. The symbol  indicates acceleration of the reaction.
indicates acceleration of the reaction.
Clinical features

Disease Severity
Disease Parameter
Severe
Moderate
Mild
Clotting factor level (VIII or IX)
Less than 1% of normal
Between 1% and 5% of normal
Between 6% and 24% of normal
Bleeding tendency
Can bleed with very mild injury
Can bleed with moderate injury
Can bleed with severe injury, surgery, or invasive procedures
Bleeding frequency
May bleed once or twice a week
May bleed once a month
May never have a bleeding episode
Occurrence of joint bleeding
Frequent
Less frequent
Infrequent, but can occur in response to severe injury
Overview of therapy
Pain management
Immunization
Preparations used to treat hemophilia
Factor VIII concentrates
Production methods and product safety

Type of Preparation
Factor VIII
Factor IX
Recombinant
Third generation
Advate
BeneFix
Xyntha
Second generation
Helixate FS
Kogenate FS
ReFacto
First generation
Recombinate
Plasma-Derived
Ultrapure
Hemofil-M
AlphaNine SD
Monoclate-P
Bebulin VH‡
Wilate*,†
Mononine
Profilnine SD‡
Proplex T‡
Intermediate and high purity
Alphanate*
Koate-DVI*
Humate-P*
Plasma-derived factor VIII.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Drugs for hemophilia
Only gold members can continue reading. Log In or Register to continue
Get Clinical Tree app for offline access
